
More Information
Submitted: April 15, 2024 | Approved: May 24, 2024 | Published: May 27, 2024
How to cite this article: Behari M. A Rare Symptomatic Case of Heterozygous Cerebro-Tendinous Xanthomatosis (CTX) Treated with Urso-Deoxycholic Acid (UDCA): With Mini Review. J Neurosci Neurol Disord. 2024; 8: 057-063.
DOI: 10.29328/journal.jnnd.1001098
Copyright License: © 2024 Behari M. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: CYP27A1 mutation; Cholestenol; Cheno-deoxycholic acid; Urso-deoxycholic acid; Spino-cerebellar ataxia; Atherosclerosis; Cataract; Cholestatic jaundice

A Rare Symptomatic Case of Heterozygous Cerebro-Tendinous Xanthomatosis (CTX) Treated with Urso-Deoxycholic Acid (UDCA): With Mini Review
Madhuri Behari*
Professor & Head, Department of Neurology, Fortis Hospital, Vasant Kunj, Formerly Professor & Head, Neurology Department, All India Institute of Medical Sciences, New Delhi, India
*Address for Correspondence: Dr. Madhuri Behari, Professor & Head, Department of Neurology, Fortis Hospital, Vasant Kunj, Formerly Professor & Head, Neurology Department, All India Institute of Medical Sciences, New Delhi, India, Email: [email protected]
Cerebrotendinous Xanthamatosis (CTX) is a rare autosomal recessive disorder caused by a mutation in the CYP27A1 gene leading to impaired metabolism of cholesterol and accumulation of cholestenol and the cholesterol in various tissues such as the brain, eyes, lungs and bones and reduced formation of cheno-deoxycholic acid (CDCA). The clinical presentation is diverse, starting in the early neonatal period and progressing till adulthood unless treated early. A common neurological manifestation is a spino-cerebellar ataxia followed by spastic paraparesis. Tendon xanthoma is a classical finding that usually helps in clinching the diagnosis but may not be present in all cases. Brain MRI also reveals characteristic abnormalities with cerebellar atrophy and hyper-intensities in the dentate nucleus and surrounding cerebellar white matter on T1weighted images. It is a rare cause of treatable ataxia in young individuals. Treatment is by replacement by CDCA or Urso-deoxycholic acid (UDCA). Supplemented with statins these individuals also have premature atherosclerosis causing death due to athero-sclerotic coronary artery disease. Here a rare case of symptomatic heterologous CYP27A1 mutation is reported with syndrome of spino-cerebellar ataxia treated with UDCA.
Cerebro-tendinous Xanthamatosis (CTX) is a slowly progressive, rare autosomal recessive disorder of fat metabolism with variable presentations. The symptoms and signs increase with age in untreated patients. It is a rare cause of treatable cerebellar ataxia in young persons and should not be missed. The disease occurs due to a mutation in the CYP27A1 gene [1], preventing cholesterol from being converted to cheno-deoxycholic acid (CDCA). It is recommended that treatment should be started early as soon as the diagnosis is made because delay results in incomplete recovery or no recovery. Screening for CTX should be done in infants and children with cholestatic jaundice, cataracts, and chronic diarrhoea to save these individuals from disabling neuro-psychiatric problems.
A 36-year-old Muslim man from Bihar (Eastern state in India), born of non-con-consanguineous parents was first seen with poor balance for 3 months - 4 months and unclear speech for two months. His birth was normal and so were his milestones. In his early childhood, he had frequent diarrhoea and was of a lean build as compared to his three younger brothers. He had bilateral cataracts since the age of 15 years, not impairing vision. There was no history of any psychiatric illness or tremors. Due to mental sub-normality, he could not study beyond class VIII in school. His mother has had high cholesterol and type 2 diabetes mellitus since the age of 50 years. Out of ten of his mother’s siblings, eight have high cholesterol levels and five have had acute myocardial infarction at ages ranging from 40 to 48 years. On general physical examination, he had 10 cm. x 8 cm. firm, fixed non-tender swellings on Achilles’ tendon bilaterally (Figure 1). His higher mental examination showed an MMSE score of 24/30. He had bilateral cataracts, cranial nerves were normal without nystagmus. Bilateral cerebellar signs were present with the abnormal finger-nose test, dysdiadochokinesis, and ataxic spastic gait. Power in all muscles was normal with exaggerated deep tendon jerks and bilateral extensor plantar response. CBC, LFT, and KFT were normal. Lipid profile showed total cholesterol 190 mg/dL, triglycerides 107 mg/dL, HDL cholesterol 70 mg/dL, LDL cholesterol 106 mg/dL, non-HDL cholesterol 104 mg/dL, VLDL 20.7 mg/dL. Acid steroids and bile alcohols in urine could not be evaluated. A serum cholestenol test also could not be done.
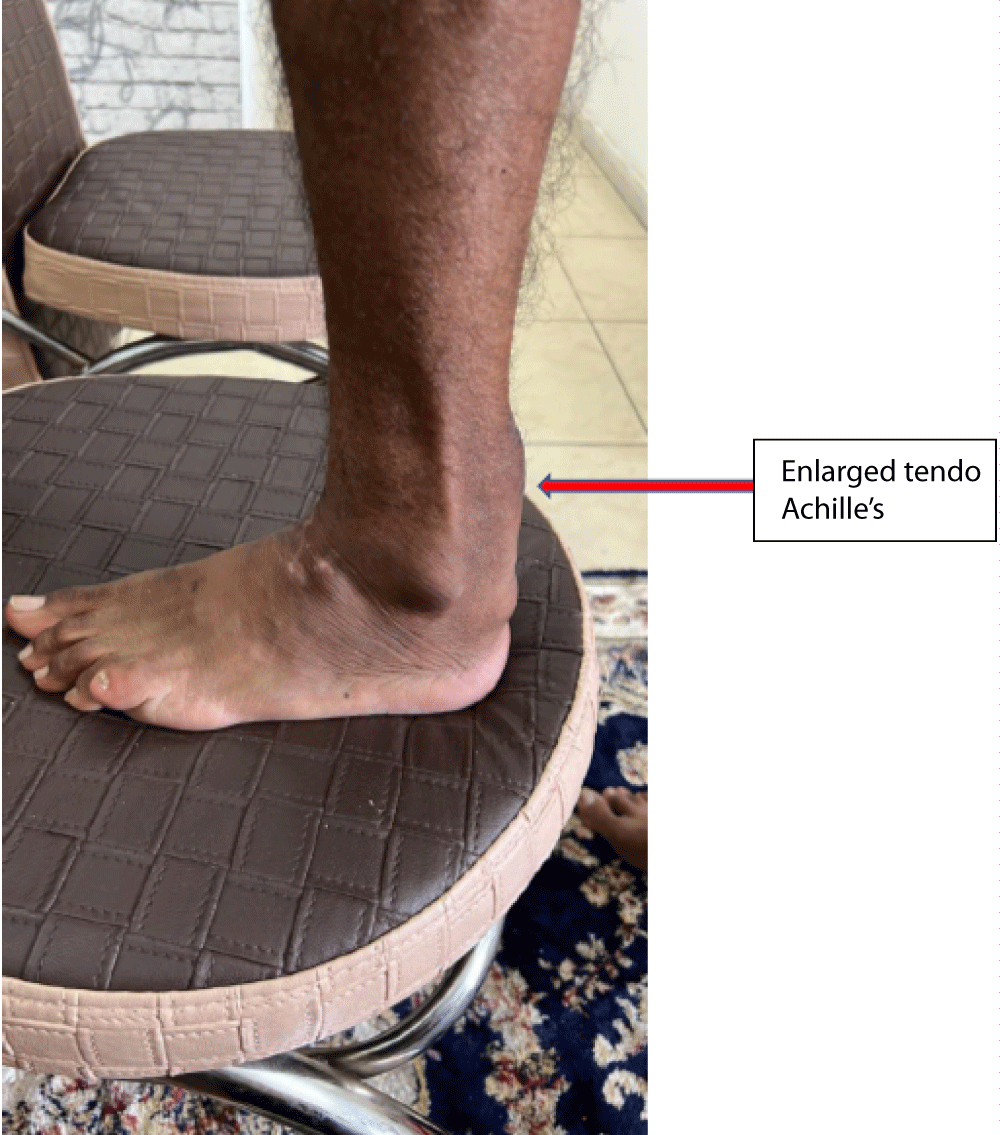
Figure 1: Showing swelling on Achilles tendon of the patient.
His MRI brain showed hypo-intensities in bilateral cerebellar hemispheres and dentate nuclei on T1 weighted images and T2 weighted images showed signal hyper-intensities on T2 weighted images (Figures 2a,2b). Cerebral hemispheres and brain stem were unremarkable. A biopsy of swelling on the Achilles tendon showed foam cells with scattered giant cells suggestive of Xanthoma. Genetic test performed showed mutation in CYP27A1: Location: chr2: 219677022: c c.526del: p.Asp176MetfsTer6: Depth: 142. It was a heterozygous frame-shift variant (c.526del) in exon 3 that resulted in the premature termination of the protein (p.Asp176MetfsTer6. On the basis of clinical, imaging, pathological, and genetic tests basis he was diagnosed with Cerebro-Tendinous Xanthomatosis (CTX) and started on Ursodecholic acid (UDCA) 250 mg thrice daily. He improved remarkably over the next 3 months, but due to the non-availability of UDCA in his country of residence, he stopped it for about a month. When seen at this time he was severely spastic with almost whispering speech and dependent on mobility and self-care. It was reinforced to him and his family the importance of continuing therapy and a statin was added too due to a poor family history of athero-sclerotic coronary artery disease. After this, he still has to come for a follow-up. The genetic test of the mother could not be done due to financial constraints.
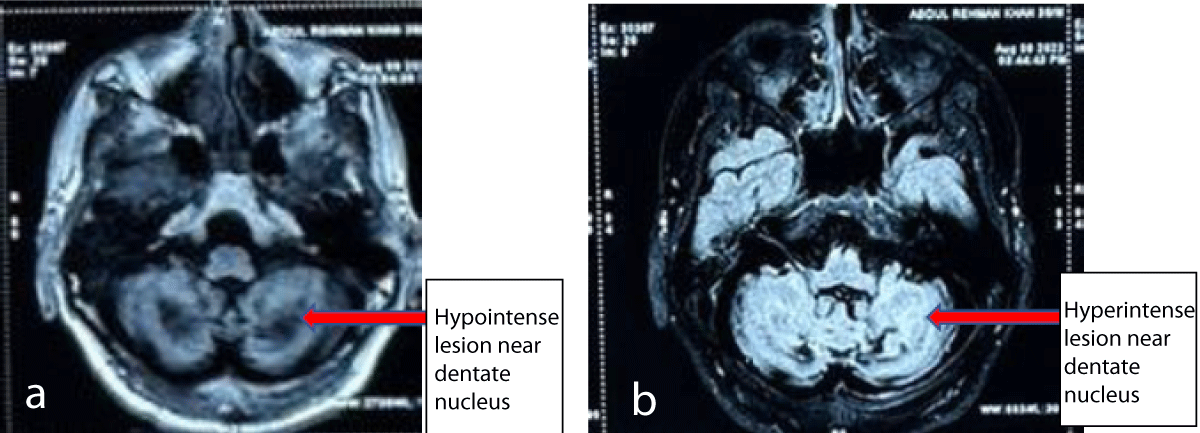
Figure 2: Non-contrast MRI of brain T1 image showing hypointense lesion in bilateral cerebellar hemispheres surrounding dentate nuclei (a) and T2 images showing hyperintense lesions in the same area (b).
The case presented here is special in several ways. The patient presented as a case of spino-cerebellar ataxia though at a much younger age, and the presence of tendon xanthoma on the Achilles’ tendon gave away the diagnosis. He had a heterozygous mutation in CYP27A1 and was still symptomatic, defying the rules of genetics that autosomal recessive disorders are symptomatic if homozygous and heterozygous are carriers. However, there is an observation by Federico, et al. of symptomatic heterozygous cases, though they did not report their case [2]. Gene mutation observed in this case is the same as reported by Dutta, et al. in Eastern Indian and Surinamese patients [i.e. CYP27A1 gene (c.526delG)], and consider it the founder mutation [3]. We treated our patient with urso-deoxy cholic acid (UDCA) as cheno-deoxycholic acid (CDCA) is not available in India. Though CDCA is the recommended treatment of CTX, we used UDCA since it has been used earlier in the USA, Australia, Bangladesh, India and Japan [3-7]. CDCA has side effects in the form of varying degrees of hepatic injury [8-11], but UDCA has been shown to be safe in individuals with gastrostomy to prevent gallstones, and other disorders [12].
Since the first description of the Cerebrotendinous Xanthomatosis (CTX) patient by van Bogaert, et al. in 1937 [1], more than 400 patients have been described worldwide [13]. Cerebrotendinous Xanthomatosis (CTX) is a relatively rare disorder described in Jews of North African descent, a group of Japanese cases, Arab Druze communities, and in Italy, The Netherlands, and Pakistan. It is a rare autosomal-recessive lipid storage disease caused by deficiency of the mitochondrial cytochrome P 450 enzyme, sterol 27-hydroxylase (CYP27A1) due to mutations in the CYP27A1 gene [1], preventing cholesterol from being converted to cheno-deoxycholic acid (CDCA). It was in 1968 that Menkes, et al. identified the storage material as cholestenol [14]. Cholestenol (a derivative of cholesterol) gets deposited in nerve cells and membranes, resulting in damage to the brain, spinal cord, tendons, lens of the eye, and arteries. People with CTX may die prematurely because of progressive neurological deterioration or coronary artery disease. Delayed diagnosis of this disease may have devastating effects on a patient’s life. Long-term treatment with CDCA significantly affects the neurophysiology and improves the disease outcome.
Recent estimates of incidence of CTX range from about 1:135,000 to 1:460,000 in people of European origin, about 1 in 70,000 in people of Asian origin, 1:263,222-1:468,624 in Africans, 1:64,247- 1:64,712 in East Asians, and 1:36,072-1:75,601 in South Asians [15,16] in an isolated Israeli Druze community a carrier frequency of 1:11 for the deleterious c.355delC variant was determined, leading to an estimated prevalence of CTX at 1:440 individuals. A founder mutation in the CYP27A1 gene (c.526delG) has been reported in the Eastern Indian and Surinamese populations [3]. It is believed that the reported estimates of prevalence may be an underestimate, as the disease gets missed due to diverse presentation even in the same family and its rarity.
CTX is associated with considerable variability in clinical manifestations among patients and even within the same family [17]. Clinical presentations vary with the age of patients, sometimes symptoms present in infancy or childhood are ignored and diagnosis is clinched only when neurological signs make their appearance (Table 1). The clinical course is relentlessly progressive unless treated. For this reason, if treatment is delayed it leads to incomplete improvement. The first symptom occurs as cholestatic jaundice in neonates and in some cases during intra-uterine life. Mutations in the CYP27A1 gene lead to cholestasis which may affect nuclear receptors such as farnesoid X receptor (FXR) [18]. Marked reduction of CDCA in CTX leads to decreased activation of FXR, which results in reduced expression of the bile salt export pump, causing a decrease in canalicular bile salt transportation [18,19]. It causes blockage of biliary canaliculi by bile salts and cholesterol crystals. This leads to jaundice for brief periods or even chronic cirrhosis. Childhood is marked by the presence of chronic diarrhoea with difficulty in gaining weight. Later around puberty, children develop cataracts, which may or may not cause visual impairment, depending on whether they are irregular cortical opacities, anterior polar cataracts, or dense posterior sub-capsular cataracts [20]. In the eye, optic atrophy has been reported but later in life. During childhood and teenage several neuropsychiatric symptoms like mental sub-normality, depression, mood swings, aggression disability, ADHD, suicidal tendencies, etc. make their appearance [17,21,22]. Tendon xanthomas usually appear in the second decade. Though most typical on Achilles’ tendon, xanthomas are also reported on other tendons like the patella, extensors of the finger, and at the elbow. In a small percentage, xanthomas may not be present at all, though they are the hallmark of the disease. Xanthomas have also been observed in the brain, bones, and lungs [23].
| Table 1: Shows symptoms according to the age of patients. | |||
| S. No. | Age | Clinical Feature | % of patients |
| 1. | Infancy | Cholestatic Jaundice | 40 |
| 2. | Childhood | Diahorrea | 89 |
| 3. | Teenage | Low intelligence, learning disability, slow learning | 60 |
| Psychiatric, depression, aggressive behaviour, suicidal tendency, ADHD, oppositional defiant disorder | 44 | ||
| 4. | 2nd - 3rd Decade | Difficulty in walking, falls, spastic gait. Staggering gait, cerebellar signs, | 36 |
| Tendon xanthoma | 78 | ||
| spasticity | 64 | ||
| 5. | Adulthood | Epilepsy | 33 |
| Peripheral neuropathy | 70 | ||
| Parkinsonism, slowness, oro-mandibular dystonia | 9 | ||
| Coronary artery disease | 25 | ||
| Osteopenia | 67 | ||
During the second or third decade, neurological symptoms appear in the form of difficulty in walking, staggering gait, frequent falls, ataxia, spasticity, peripheral neuropathy, and bone abnormalities leading to skeletal deformities like pes-cavus and scoliosis. Typical MRI findings in CTX are cerebellar lesions, particularly those in and surrounding the dentate nuclei [24]. Rarely, do some patients develop autonomic disturbance and extrapyramidal features with tremors, slowness, soft speech, tachylalia (fast speech) [17,21,22,25,26], and dystonia. Epilepsy occurs in about one-third of patients. A rare spinal form is also described with a relatively better prognosis characterised by spasticity and posterior column involvement resembling sub-acute combined degeneration of the cord (SACD) [27-29]. In the spinal form, an MRI of the spinal cord may or may not show hypointensity on T1 weighted images in lateral and posterior columns. In a small percentage, the MRI may not show any abnormalities. Spinal form can develop at any stage of life.
Apart from the above, other non-neurological features consist of:
- Skeletal system: Osteoporosis and osteopenia seen in about 67% of patients occur due to poor absorption of vitamin D and Calcium due to a lack of bile acids and bile salts [30,31]. It leads to skeletal deformities like scoliosis, kyphosis, pectus excavatum, pes equino-varus, and pes cavus [32]. In addition, muscle disorders like myopathies and neurogenic muscle involvement due to peripheral neuropathy are also seen [33,34].
- Cardiovascular system: Premature atherosclerosis resulting in myocardial infarction has been reported in young individuals as young as in their fourth decade [35,36] which may cause premature death [36], aortic aneurysm, carotid atherosclerotic lesions, and thickening of the interatrial septum compatible with lymphomatous hypertrophy [37-39].
- Lungs: In the lungs infiltration of alveolar septa with lymphomatous foamy cells leading to breathing difficulty has been observed.
Diagnosis
Diagnostic criteria was proposed by Sekijima, et al. Essentially, it consists of seven symptoms, namely tendon xanthoma, progressive neuropsychiatric dysfunction or mental retardation, juvenile cataract, juvenile coronary artery disease, chronic unexplained diarrhea, juvenile osteoporosis, prolonged neonatal cholestasis; Biochemical abnormality consisting of elevated serum cholesterol level; and genetic test showing a pathogenic mutation in CYP27A1 gene (homozygosity or compound heterozygosity). There are three diagnostic categories: definite, probable, and possible.
Differential diagnoses are:
- Familial hypercholesterolemia
- Sitosterolemia
- Obstructive biliary tract disease
- Hypothyroidism
Diagnostic categories are:
- Definite: Presence of at least one of the symptoms+ biochemical criteria + genetic + differential diagnoses should be excluded
- Probable: Presence of at least one symptom+ Biochemical + Genetic
- Possible: Presence of at least one symptom + biochemical
The index of suspicion for the diagnosis of CTX should be high in all cases of:
- Typical findings on Magnetic resonance imaging (MRI) of the brain showing symmetrical high-signal lesions in the globus pallidus and in the deep cerebellar white matter, together with low signal lesions in the dentate nucleus on T2-weighted MRI [40,41]. Once these lesions are found, they are often stable in time [42].
- All patients below 30 years of age who have cataracts should be screened for CTX, especially if they also have chronic diarrhea, tendon xanthomas, and/or neuropsychiatric symptoms [43].
- The presence of tendon xanthomas is not mandatory for a diagnosis of CTX and the presence of two of the four clinical features of premature cataracts, intractable diarrhea, and progressive neurological signs and symptoms require biochemical screening for CTX [44].
- Asymptomatic siblings of patients should be screened for CTX as the sibling can be completely cured with a good prognosis.
Management
Early diagnosis is the key to the successful management of CTX. This requires a high index of suspicion esp. in children with neonatal jaundice, juvenile cataracts, and childhood diarrhoea along with neuro-psychiatric symptoms. The presence of xanthomas is not essential for diagnosis. Replacement treatment with Chenodeoxycholic Acid (CDCA) in the early stage of the disease has been reported to improve or even prevent clinical symptoms of CTX [45,46]; however, after significant neurological pathology is established, the effect of the treatment is limited and deterioration of clinical manifestations may continue to progress [11,43,47,48]. Therefore, it is crucial to treat CTX patients at the initial stage of the disease. The age of diagnosis and initiation of treatment with CDCA correlates with the prognosis of patients with CTX [11,48]. Early treatment with CDCA in pre-symptomatic individuals appears to prevent clinical manifestations. In a study, authors treated two siblings with CTX with CDCA since ages of 2 and 7 years respectively who remained asymptomatic and did not develop any neurological manifestations even after 14 years of follow-up [50]. These findings strongly suggest that early diagnosis and treatment are crucial in a better prognosis of CTX. However, generally, an average delay of diagnosis and subsequent treatment is observed in retrospective cohort studies to the tune of 15 years - 25 years [17,21,22,47].
The recommended dose of CDCA treatment is 750 mg/day divided into three doses for adults and 15 mg/kg/day for children in three divided doses given orally [44,45]. During treatment with CDCA, approximately 9% of patients require dose adjustment owing to drug-induced liver damage [42]. Hence, clinical and laboratory monitoring and dosage adjustment for CDCA are essential in the treatment of CTX, especially in infants and young children [11,42]. CDCA has been shown to cause significant liver damage and even hepatic failure. CDCA was initially preferred to cholic acid because it was more effective in reducing cholesterol 7α-hydroxylase and has a stronger negative feedback effect on it [49,50]. Therefore, due to the hepatotoxicity of CDCA, cholic acid is considered the safer option in CTX, especially in infants [51]. Mandia, et al. reported the efficacy of cholic acid in adult patients with CTX, including individuals whose CDCA treatment was discontinued due to the non-availability of CDCA [50]. Treatment with cholic acid has been shown to significantly reduce cholestanol levels in all patients but also leads to improvement or stabilization of systemic and/or neurological manifestations [50]. Due to the non-availability or non-approval of CDCA in CTX, Urso-deoxycholic acid (UDCA) has been used in some countries like the USA, Japan, and India [3,6,12]. UDCA is free from hepatotoxicity and according to recent reports, is efficacious even after long use, despite some contrary reports to this effect in the past [52]. The dose of UDCA is 300 mg given three times a day orally.
It is suggested that at the first diagnosis, certain tests should be done in patients with CTX (Table 2), and during therapy hepatic function should be performed periodically.
| Table 2: Recommended tests in CTX during therapy with CDCA. | ||
| S. No. | Problems | Tests |
| 1. | Elevated cholestanol level | Lab testing of lipids including plasma cholestanol level |
| 2. | Peripheral neuropathy | EMG & NCV studies |
| 3. | Cardiologic issues | EKG & echocardiogram |
| 4. | Osteoporosis | Bone density study |
| 5. | Cataracts | Ophthalmologic evaluation |
| 6. | Neurologic & behavioral concerns | Neurological and psychiatric evaluation |
Supportive treatment
All patients should receive combination therapy with CDCA and HMG-CoA reductase inhibitors (statins) because of their synergistic effects on serum cholestanol and urine bile alcohol levels [52,53]. However, some report the absence of this synergistic effect [54]. Inhibitors of HMG-CoA reductase alone or in combination with CDCA are also effective in decreasing cholestanol concentration and improving clinical signs and atherosclerotic coronary and carotid artery disease.
Genetic counselling
Genetic counselling is required for the parents, patient, and siblings of the patient. They also have to be screened for pre-symptomatic disease and treated if found to be homozygous or compound heterozygous for CTX.
The case presented here is a rare case of CTX with a heterozygous mutation on CYP27A1 who developed typical clinical features of the disorder. The following special features that this case demonstrates are:
- This case is a rare case who is heterozygous for CYP 27A1 mutation and also showed classical symptoms of CTX, though Federico, et al. have also observed some patients who are heterozygous and symptomatic [2].
- Of all hereditary spino-cerebellar ataxia, CTX is a rare type that is completely treatable if treatment is started early.
- Due to diverse clinical manifestations and its rarity, the diagnosis of CTX is likely to be missed in many cases.
- The index of suspicion for the diagnosis of CTX should be high in children and young adults with chronic diarrhoea, cholestatic jaundice, premature cataract, psychiatric symptoms, atherosclerosis with coronary artery involvement, early death, and progressive neurological symptoms. Either all of them or any two or more of these should arouse suspicion of CTX.
- Clinical presentations are diverse among patients and even within the same family. Even neurologically, the symptoms are variable. Common ones being cerebellar ataxia, spastic paraparesis. Others are neuropathy, optic atrophy, cognitive problems, epilepsy, and extra-pyramidal and autonomic system involvement.
- It is a systemic disorder with non-neuropsychiatric symptoms like osteoporosis, joint problems, cardiac involvement, aortic aneurysm, lung involvement, and liver involvement.
Patient consent statement
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. The participant and his legally authorized representative provided written informed consent before entering the study in compliance with the applicable local regulations.
- Cali JJ, Hsieh CL, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem. 1991 Apr 25;266(12):7779-83. PMID: 2019602; PMCID: PMC4449724.
- Federico A, Gallus GN. Cerebrotendinous Xanthomatosis. 2003 Jul 16 [Updated 2022 Mar 17]. In: Adam MP, Mirzaa GM, Pagon RA, et al. [Eds.] GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Bookshelf URL: https://www.ncbi.nlm.nih.gov/books/
- Dutta AK, Danda S, Muthusamy K, Alexander M, Sudhakar SV, Hansdak S, Bandyopadhyay R, Bakhya Shree GB, Rekha L. Cerebrotendinous xanthomatosis: Possibility of founder mutation in CYP27A1 gene (c.526delG) in Eastern Indian and Surinamese population. Mol Genet Metab Rep. 2015 Mar 23;3:33-5. doi: 10.1016/j.ymgmr.2015.03.002. PMID: 26937392; PMCID: PMC4750635.
- Mirzanli C, Esenyel CZ, Ozturk K, Baris A, Imren Y. Cerebrotendinous xanthomatosis presenting with bilateral achilles tendon xanthomata: a case report. J Am Podiatr Med Assoc. 2013 Mar-Apr;103(2):152-5. doi: 10.7547/1030152. PMID: 23536508.
- Chowdhury G, Kayasthagir PK, Chowdhury A. Cerebrotendinous xanthomatosis: case report on a rare genetic case. JCMCTA. 2017;28(1):72-75.
- Shaji B, Srikumar B, Ramachandran D. A Preventable Ataxia: Cerebrotendinous Xanthomatosis. Ann Indian Acad Neurol. 2019 Oct-Dec;22(4):493-496. doi: 10.4103/aian.AIAN_126_18. Epub 2019 Oct 25. PMID: 31736580; PMCID: PMC6839296.
- Kimura S, Beppu T, Kugai N, Koide Y, Fujita T, Iida K, Yamashita N, Yamashita K, Seyama Y. A case of cerebrotendinous xanthomatosis: effects of ursodeoxycholic acid administration on serum bile acids and cholestanol. Jpn J Med. 1982 Jul;21(3):210-5. doi: 10.2169/internalmedicine1962.21.210. PMID: 7143816.
- Abe R, Sekijima Y, Kinoshita T, Yoshinaga T, Koyama S, Kato T, Ikeda SI. Spinal form cerebrotendinous xanthomatosis patient with long spinal cord lesion. J Spinal Cord Med. 2016 Nov;39(6):726-729. doi: 10.1179/1079026815Z.000000000409. Epub 2016 Feb 25. PMID: 25941960; PMCID: PMC5137569.
- Waterreus RJ, Koopman BJ, Wolthers BG, Oosterhuis HJ. Cerebrotendinous xanthomatosis (CTX): a clinical survey of the patient population in The Netherlands. Clin Neurol Neurosurg. 1987;89(3):169-75. doi: 10.1016/s0303-8467(87)80050-1. PMID: 3665290.
- Kuriyama M, Tokimura Y, Fujiyama J, Utatsu Y, Osame M. Treatment of cerebrotendinous xanthomatosis: effects of chenodeoxycholic acid, pravastatin, and combined use. J Neurol Sci. 1994 Aug;125(1):22-8. doi: 10.1016/0022-510x(94)90237-2. PMID: 7964884.
- Huidekoper HH, Vaz FM, Verrips A, Bosch AM. Hepatotoxicity due to chenodeoxycholic acid supplementation in an infant with cerebrotendinous xanthomatosis: implications for treatment. Eur J Pediatr. 2016 Jan;175(1):143-6. doi: 10.1007/s00431-015-2584-7. Epub 2015 Jul 10. PMID: 26156051; PMCID: PMC4709371.
- Lee SH, Jang DK, Yoo MW, Hwang SH, Ryu SY, Kwon OK, Hur H, Man Yoon H, Eom BW, Ahn HS, Son T, Song KY, Lee HH, Choi MG, An JY, Lee SI, Lee KH, Ahn S, Park YS, Park DJ; Efficacy and Safety of DWJ1319 in the Prevention of Gallstone Formation after Gastrectomy in Patient with Gastric Cancer: A Multicenter, Randomized, Double-blind, Placebo-controlled Study (PEGASUS-D) Group. Efficacy and Safety of Ursodeoxycholic Acid for the Prevention of Gallstone Formation After Gastrectomy in Patients With Gastric Cancer: The PEGASUS-D Randomized Clinical Trial. JAMA Surg. 2020 Aug 1;155(8):703-711. doi: 10.1001/jamasurg.2020.1501. PMID: 32584935; PMCID: PMC7301302.
- Stelten BML, Dotti MT, Verrips A, Elibol B, Falik-Zaccai TC, Hanman K, Mignarri A, Sithole B, Steiner RD, Verma S, Yahalom G, Zubarioglu T, Mochel F, Federico A. Expert opinion on diagnosing, treating and managing patients with cerebrotendinous xanthomatosis (CTX): a modified Delphi study. Orphanet J Rare Dis. 2021; 16:353.
- Menkes JH, Schimschock JR, Swanson PD. Cerebrotendinous xanthomatosis. The storage of cholestanol within the nervous system. Arch Neurol. 1968 Jul;19(1):47-53. doi: 10.1001/archneur.1968.00480010065004. PMID: 5676919.
- Salen G, Steiner RD. Epidemiology, diagnosis, and treatment of Cerebrotendinous Xanthomatosis (CTX). J Inherit Metab Dis. 2017; 40: 771–81. doi: 10.1007/s10545-017-0093-8
- Appadurai V, DeBarber A, Chiang PW, Patel SB, Steiner RD, Tyler C, Bonnen PE. Apparent underdiagnosis of Cerebrotendinous Xanthomatosis revealed by analysis of ~60,000 human exomes. Mol Genet Metab. 2015 Dec;116(4):298-304. doi: 10.1016/j.ymgme.2015.10.010. Epub 2015 Oct 26. PMID: 26643207; PMCID: PMC4767010.
- Verrips A, Hoefsloot LH, Steenbergen GC, Theelen JP, Wevers RA, Gabreëls FJ, van Engelen BG, van den Heuvel LP. Clinical and molecular genetic characteristics of patients with cerebrotendinous xanthomatosis. Brain. 2000 May;123 ( Pt 5):908-19. doi: 10.1093/brain/123.5.908. PMID: 10775536.
- Edwards PA, Kast HR, Anisfeld AM. BAREing it all: the adoption of LXR and FXR and their roles in lipid homeostasis. J Lipid Res. 2002 Jan;43(1):2-12. PMID: 11792716.
- von Bahr S, Björkhem I, Van’t Hooft F, Alvelius G, Nemeth A, Sjövall J, Fischler B. Mutation in the sterol 27-hydroxylase gene associated with fatal cholestasis in infancy. J Pediatr Gastroenterol Nutr. 2005 Apr;40(4):481-6. doi: 10.1097/01.mpg.0000150419.23031.2a. Erratum in: J Pediatr Gastroenterol Nutr. 2005 Jul;41(1):144. PMID: 15795599.
- Cruysberg JR, Wevers RA, van Engelen BG, Pinckers A, van Spreeken A, Tolboom JJ. Ocular and systemic manifestations of cerebrotendinous xanthomatosis. Am J Ophthalmol. 1995 Nov;120(5):597-604. doi: 10.1016/s0002-9394(14)72206-8. PMID: 7485361.
- Mignarri A, Gallus GN, Dotti MT, Federico A. A suspicion index for early diagnosis and treatment of cerebrotendinous xanthomatosis. J Inherit Metab Dis. 2014 May;37(3):421-9. doi: 10.1007/s10545-013-9674-3. Epub 2014 Jan 18. PMID: 24442603.
- Sekijima Y, Koyama S, Yoshinaga T, Koinuma M, Inaba Y. Nationwide survey on cerebrotendinous xanthomatosis in Japan. J Hum Genet. 2018 Mar;63(3):271-280. doi: 10.1038/s10038-017-0389-4. Epub 2018 Jan 10. PMID: 29321515.
- Brienza M, Fiermonte G, Cambieri C, Mignarri A, Dotti MT, Fiorelli M. Enlarging brain xanthomas in a patient with cerebrotendinous xanthomatosis. J Inherit Metab Dis. 2015 Sep;38(5):981-2. doi: 10.1007/s10545-014-9805-5. Epub 2015 Jan 8. PMID: 25567502.
- Nie S, Chen G, Cao X, Zhang Y. Cerebrotendinous Xanthomatosis: a comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J Rare Dis. 2014; 9: 179. doi: 10.1186/s13023-014-0179-4.
- Wong JC, Walsh K, Hayden D, Eichler FS. Natural history of neurological abnormalities in cerebrotendinous xanthomatosis. J Inherit Metab Dis. 2018 Jul;41(4):647-656. doi: 10.1007/s10545-018-0152-9. Epub 2018 Feb 26. PMID: 29484516.
- Bartholdi D, Zumsteg D, Verrips A, Wevers RA, Sistermans E, Hess K, Jung HH. Spinal phenotype of cerebrotendinous xanthomatosis--a pitfall in the diagnosis of multiple sclerosis. J Neurol. 2004 Jan;251(1):105-7. doi: 10.1007/s00415-004-0221-x. PMID: 14999499.
- Mutlu D, Tuncer A, Gocmen R, Yalcin-Cakmakli G, Saygı S, Elibol B: Diagnostic challenge: A case of late- Bartholdi D, Zumsteg D, Verrips A, Wevers RA, Sistermans E, Hess K, Jung HH: Spinal phenotype of cerebrotendinous xanthomatosis--a pitfall in the diagnosis of multiple sclerosis. J Neurol, 2004; 251: 105-107
- Dotti MT, Lütjohann D, von Bergmann K, Federico A. Normalisation of serum cholestanol concentration in a patient with cerebrotendinous xanthomatosis by combined treatment with chenodeoxycholic acid, simvastatin and LDL apheresis. Neurol Sci. 2004 Oct;25(4):185-91. doi: 10.1007/s10072-004-0320-6. PMID: 15549503.
- Koopman BJ, Wolthers BG, van der Molen JC, Waterreus RJ. Bile acid therapies applied to patients suffering from cerebrotendinous xanthomatosis. Clin Chim Acta. 1985 Oct 31;152(1-2):115-22. doi: 10.1016/0009-8981(85)90182-2. PMID: 4053393.
- Zubarioglu T, Kiykim E, Yesil G, Demircioglu D, Cansever MS, Yalcinkaya C, Aktuglu-Zeybek C. Early diagnosed cerebrotendinous xanthomatosis patients: clinical, neuroradiological characteristics and therapy results of a single center from Turkey. Acta Neurol Belg. 2019 Sep;119(3):343-350. doi: 10.1007/s13760-017-0851-2. Epub 2017 Oct 22. PMID: 29058268.
- Lumbreras S, Ricobaraza A, Baila-Rueda L, Gonzalez-Aparicio M, Mora-Jimenez L, Uriarte I, Bunuales M, Avila MA, Monte MJ, Marin JJG, Cenarro A, Gonzalez-Aseguinolaza G, Hernandez-Alcoceba R. Gene supplementation of CYP27A1 in the liver restores bile acid metabolism in a mouse model of cerebrotendinous xanthomatosis. Mol Ther Methods Clin Dev. 2021 Jul 21;22:210-221. doi: 10.1016/j.omtm.2021.07.002. PMID: 34485606; PMCID: PMC8399082.
- Verrips A, van Engelen BG, ter Laak H, Gabreëls-Festen A, Janssen A, Zwarts M, Wevers RA, Gabreëls FJ. Cerebrotendinous xanthomatosis. Controversies about nerve and muscle: observations in ten patients. Neuromuscul Disord. 2000 Aug;10(6):407-14. doi: 10.1016/s0960-8966(00)00112-7. PMID: 10899446.
- Federico A, Dotti MT, Volpi N. Muscle mitochondrial changes in cerebrotendinous xanthomatosis. Ann Neurol. 1991 Nov;30(5):734-5. doi: 10.1002/ana.410300517. PMID: 1763899.
- Kuriyama M, Fujiyama J, Yoshidome H, Takenaga S, Matsumuro K, Kasama T, Fukuda K, Kuramoto T, Hoshita T, Seyama Y, et al. Cerebrotendinous xanthomatosis: clinical and biochemical evaluation of eight patients and review of the literature. J Neurol Sci. 1991 Apr;102(2):225-32. doi: 10.1016/0022-510x(91)90073-g. PMID: 2072121.
- Valdivielso P, Calandra S, Durán JC, Garuti R, Herrera E, González P. Coronary heart disease in a patient with cerebrotendinous xanthomatosis. J Intern Med. 2004 Jun;255(6):680-3. doi: 10.1111/j.1365-2796.2004.01316.x. PMID: 15147532.
- Souto MJS, Almeida-Santos MA, Ferreira EJP, Gonçalves LFG, Oliveira JLM, Sousa ACS. Spontaneous Coronary Artery Dissection in a Patient with Cerebrotendinous Xanthomatosis. Arq Bras Cardiol. 2020 Apr;115(1 suppl 1):18-21. English, Portuguese. doi: 10.36660/abc.20190456. PMID: 32935759; PMCID: PMC8386949.
- Potkin BN, Hoeg JM, Connor WE, Salen G, Quyyumi AA, Brush JE Jr, Roberts WC, Brewer HB Jr: Aneurysmal coronary artery disease in cerebrotendinous xanthomatosis. Am J Cardiol, 1988; 61: 1150-1152.
- Dotti MT, Mondillo S, Plewnia K, Agricola E, Federico A. Cerebrotendinous xanthomatosis: evidence of lipomatous hypertrophy of the atrial septum. J Neurol. 1998 Nov;245(11):723-6. doi: 10.1007/s004150050274. PMID: 9808240.
- Saute JA, Giugliani R, Merkens LS, Chiang JPW, DeBarber AE, de Souza CFM. Look carefully to the heels! a potentially treatable cause of spastic paraplegia. J Inherit Metab Dis. 2015; 38: 363–4. doi: 10.1007/s10545-014-9745-0
- Burguez D, Polese-Bonatto M, Scudeiro LAJ, Björkhem I, Schöls L, Jardim LB, Matte U, Saraiva-Pereira ML, Siebert M, Saute JAM. Clinical and molecular characterization of hereditary spastic paraplegias: A next-generation sequencing panel approach. J Neurol Sci. 2017 Dec 15;383:18-25. doi: 10.1016/j.jns.2017.10.010. Epub 2017 Oct 10. PMID: 29246610.
- Atallah I, Millán DS, Benoît W, Campos-Xavier B, Superti-Furga A, Tran C. Spinal cerebrotendinous xanthomatosis: A case report and literature review. Mol Genet Metab Rep. 2021 Feb 3;26:100719. doi: 10.1016/j.ymgmr.2021.100719. PMID: 33659184; PMCID: PMC7890005.
- Duell PB, Salen G, Eichler FS, DeBarber AE, Connor SL, Casaday L, Jayadev S, Kisanuki Y, Lekprasert P, Malloy MJ, Ramdhani RA, Ziajka PE, Quinn JF, Su KG, Geller AS, Diffenderfer MR, Schaefer EJ: Diagnosis, treatment, and clinical outcomes in 43 cases with cerebrotendinous xanthomatosis. J Clin Lipidol, 2018; 12: 1169-1178.
- Jain RS, Sannegowda RB, Agrawal A, Hemrajani D, Jain R, Mathur T. ‘Hot cross bun’ sign in a case of cerebrotendinous xanthomatosis: a rare neuroimaging observation. BMJ Case Rep. 2013; doi: 10.1136/bcr- 2012-006641
- Berginer VM, Salen G, Shefer S. Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid. N Engl J Med. 1984 Dec 27;311(26):1649-52. doi: 10.1056/NEJM198412273112601. PMID: 6504105.
- van Heijst AF, Verrips A, Wevers RA, Cruysberg JR, Renier WO, Tolboom JJ. Treatment and follow-up of children with cerebrotendinous xanthomatosis. Eur J Pediatr. 1998 Apr;157(4):313-6. doi: 10.1007/s004310050818. PMID: 9578968.
- Pilo-de-la-Fuente B, Jimenez-Escrig A, Lorenzo JR, Pardo J, Arias M, Ares-Luque A, Duarte J, Muñiz-Pérez S, Sobrido MJ. Cerebrotendinous xanthomatosis in Spain: clinical, prognostic, and genetic survey. Eur J Neurol. 2011 Oct;18(10):1203-11. doi: 10.1111/j.1468-1331.2011. 03439.x. Epub 2011 Jun 4. PMID: 21645175.
- Yahalom G, Tsabari R, Molshatzki N, Ephraty L, Cohen H, Hassin-Baer S. Neurological outcome in cerebrotendinous xanthomatosis treated with chenodeoxycholic acid: early versus late diagnosis. Clin Neuropharmacol. 2013 May-Jun;36(3):78-83. doi: 10.1097/WNF.0b013e318288076a. PMID: 23673909.
- Stelten BML, Huidekoper HH, van de Warrenburg BPC, Brilstra EH, Hollak CEM, Haak HR, Kluijtmans LAJ, Wevers RA, Verrips A. Long-term treatment effect in cerebrotendinous xanthomatosis depends on age at treatment start. Neurology. 2019 Jan 8;92(2):e83-e95. doi: 10.1212/WNL.0000000000006731. Epub 2018 Dec 7. PMID: 30530799.
- Ellis E, Axelson M, Abrahamsson A, Eggertsen G, Thörne A, Nowak G, Ericzon BG, Björkhem I, Einarsson C. Feedback regulation of bile acid synthesis in primary human hepatocytes: evidence that CDCA is the strongest inhibitor. Hepatology. 2003 Oct;38(4):930-8. doi: 10.1053/jhep.2003.50394. PMID: 14512880.
- Mandia D, Chaussenot A, Besson G, Lamari F, Castelnovo G, Curot J, Duval F, Giral P, Lecerf JM, Roland D, Pierdet H, Douillard C, Nadjar Y. Cholic acid as a treatment for cerebrotendinous xanthomatosis in adults. J Neurol. 2019 Aug;266(8):2043-2050. doi: 10.1007/s00415-019-09377-y. Epub 2019 May 21. PMID: 31115677.
- Pierre G, Setchell K, Blyth J, Preece MA, Chakrapani A, McKiernan P. Prospective treatment of cerebrotendinous xanthomatosis with cholic acid therapy. J Inherit Metab Dis. 2008 Dec;31 Suppl 2:S241-5. doi: 10.1007/s10545-008-0815-z. Epub 2008 Dec 27. PMID: 19125350.
- Kimura S, Beppu T, Kugai N, Koide Y, Fujita T, Iida K, Yamashita N, Yamashita K, Seyama Y. A case of cerebrotendinous xanthomatosis: effects of ursodeoxycholic acid administration on serum bile acids and cholestanol. Jpn J Med. 1982 Jul;21(3):210-5. doi: 10.2169/internalmedicine1962.21.210. PMID: 7143816.
- Verrips A, Wevers RA, Van Engelen BG, Keyser A, Wolthers BG, Barkhof F, Stalenhoef A, De Graaf R, Janssen-Zijlstra F, Van Spreeken A, Gabreëls FJ. Effect of simvastatin in addition to chenodeoxycholic acid in patients with cerebrotendinous xanthomatosis. Metabolism. 1999 Feb;48(2):233-8. doi: 10.1016/s0026-0495(99)90040-9. PMID: 10024088.
- Salen G, Batta AK, Tint GS, Shefer S. Comparative effects of lovastatin and chenodeoxycholic acid on plasma cholestanol levels and abnormal bile acid metabolism in cerebrotendinous xanthomatosis. Metabolism. 1994 Aug;43(8):1018-22. doi: 10.1016/0026-0495(94)90183-x. PMID: 8052141.