
More Information
Submitted: August 17, 2023 | Approved: August 23, 2023 | Published: August 24, 2023
How to cite this article: Sandoval MCG, Zúñiga EE, Toribio MGG. Update in the Understanding, Diagnosis, and Management of Sturge Weber Syndrome: Case Report. J Neurosci Neurol Disord. 2023; 7: 061-064.
DOI: 10.29328/journal.jnnd.1001080
Copyright License: © 2023 Sandoval MCG, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Sturge-Weber syndrome; Epilepsy; Encephalotrigeminal angiomatosis; Port wine birthmark; Nevus flammeus; Neurocutaneous disease; Phakomatoses; Status epilepticus

Update in the Understanding, Diagnosis, and Management of Sturge Weber Syndrome: Case Report
Mariana Catalina Garcini Sandoval1*, Enrique Espinosa Zúñiga2 and Martha Guadalupe García Toribio3
1Department of Neurology, Hospital General de México “Dr. Eduardo Liceaga”, Medical Resident of the Neurology Department, Mexico
2Department of Rheumatology, Centro Médico Nacional “20 de Noviembre”, Medical Resident of the Rheumatology Department, Mexico
3Department of Neurology, Hospital General de México “Dr. Eduardo Liceaga”, Staff at Epilepsy Clinic, Mexico
*Address for Correspondence: Mariana Catalina Garcini Sandoval, Department of Neurology, Hospital General de México “Dr. Eduardo Liceaga”, Medical Resident of the Neurology Department, Mexico, Email: [email protected]
Sturge-Weber Syndrome (SWS) is a congenital, vascular, neurocutaneous, uncommon disease associated with facial angiomas port wine birthmark (PWB) or “nevus flammeus”, cerebral vascularity alterations (leptomeningeal vascular malformation), and ocular disorders. It is the third most common neurocutaneous syndrome after neurofibromatosis and tuberous sclerosis. GNAQ R183Q is the most frequent related mutation, caused by a postzygotic, somatic, gain-of-function. 75% of patients present seizures during the first year of life, mainly focal motor seizures, with or without consciousness impairment.
We present the case of a 33-year-old female with a diagnosis of SWS, with refractory seizures that started at 4 months of age. In this admission, she presented upper and lower respiratory tract infections that culminated in a convulsive status epilepticus (CSE), the reason for which she required sedation and advanced airway management with adjustment of the anti-seizure medication (ASM). An electroencephalogram (EEG) was performed that reported epileptic activity, as well as an imaging study with data suggestive of calcification in the frontal and right parietal region, compatible with vascular malformation.
Sturge-Weber syndrome (SWS), also known as encephalotrigeminal angiomatosis, is a rare, neurocutaneous, non-inherited neurovascular disorder characterized by abnormalities in the vasculature associated with angiomas involving the face (PWB), choroid, cerebral vascularity alterations (leptomeningeal vascular malformation) and ocular disorders [1,2]. Primarily affects the brain, skin, and eyes and causes seizures, developmental delay, intellectual disability, learning disabilities, and glaucoma. The incidence is not well known and estimated to be 1 in 20,000 – 50,000 live births, this disease affects males and females equally and there is no race predilection [1,3]. The most common somatic mutation (p.Arg183Gln, p.R183Q) in the guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) gene [4,5] although GNA11 and GNB2 have also been implicated.
These patients have stroke-like episodes, due to impaired cerebral perfusion with increased risk of stroke, commonly appear as transient episodes, lasting more than 24 hours of hemiparesis or visual field defects, and are often difficult to distinguish from ongoing seizure activity or postictal Todd’s paresis, for which an important part is a treatment with aspirin, in addition to ASM [6-8]. 75% of patients present seizures during the first year of life, mainly focal motor onset seizures, with or without consciousness impairment, consisting of clonic motor, being the most commonly reported seizure type, to bilateral tonic-clonic seizures [6], 11% during the second year, and only 14% after the second year [8]. Patients can develop glaucoma, the most frequent being open-angle glaucoma, presenting symptoms such as vision loss, dilated conjunctival vessels, ocular pain, excessive tearing, and buphthalmos [9], an estimated 50% of children with SWS develop glaucoma during infancy or later in childhood [2]. Choroidal hemangioma (CH), which is a benign vascular tumor, is prevalent in 40% to 50% of patients with SWS. It is more often diffuse in nature but can be circumscribed and is usually ipsilateral to the facial PWB. It is caused by aberrant and enlarged vessels and vascular channels that cause variable clinical features including reduced visual acuity, refractive errors, scotoma, or retinal detachment. The ophthalmological examination is also recommended every 3 months during the first year and yearly afterward to monitor for glaucoma and choroidal hemangiomas [10].
This is a 33-year-old patient with a diagnosis of PWB at birth (Figure 1), in addition, seizure onset at age of 4 months of age with 3 seizure types: 1) Focal onset seizure with impaired awareness, left hemibody clonic to bilateral tonic-clonic, 2) Focal onset seizure with impaired awareness, myoclonic motor, and 3) Focal onset seizure with retained awareness, the tonic motor with a Jacksonian gait, characterized by onset in the left thoracic limb that spreads to the left hemibody. With a past medical history of delay in the milestones, accompanied by cognitive impairment. SWS was diagnosed based on clinical characteristics despite no genetic study being performed during the diagnostic protocol. The patient was seizure free for one year during childhood, adolescence, and adulthood, but experienced breakthrough seizures during their last admission. Her seizure rate average was usually 5 per month, with catamenial exacerbation and multiple admissions due to intractable epilepsy and once status epilepticus (SE). Previously under treatment with gabapentin 300-300-600 mg and phenytoin 150 mg TID. Previously under treatment with gabapentin 300-300-600 mg and phenytoin 150 mg TID. Her last admission was due to breakthrough seizures secondary to presenting fever and cough with expectoration, for which community-acquired pneumonia was diagnosed, culminating in a CSE. The symptoms did not respond after the administration of benzodiazepines and ASM, for which she required sedation, advanced airway management, and antibiotic treatment, remaining for 9 days in the intensive care unit. Subsequently, as part of the approach, a thyroid profile was performed, which reported thyroid-stimulating hormone 1.77 mUI/ml, T3 0.95 ng/ml, T4 12.56 um/dl, an electroencephalogram under sedation effect, which was reported with epileptiform activity over the right frontotemporal region and continue generalized slowing (Figure 2), and non-contrast brain computed tomography (CT) showing brain parenchyma with decreased volume in the right frontal and parietal region, with hyperdense linear lesions due to vascular malformation, right cerebellar atrophy, and gyri form calcifications are classically described as “tram-track sign”(Figure 3) (Singh & Keenaghan, 2023). Brain magnetic resonance imaging (MRI) was performed, where right cerebral atrophy, secondary frontal hyperostosis, and abnormal capillary vessel on the right cerebral convexity were observed (Figure 4).
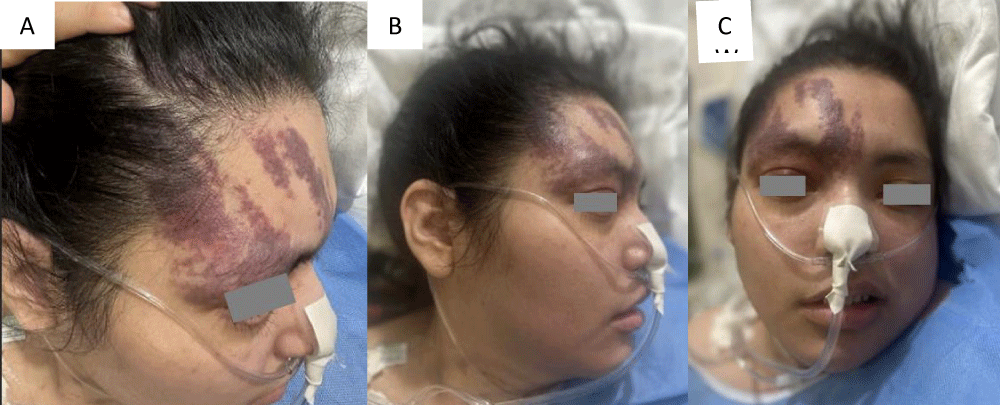
Figure 1: PWB, Type I according to the Roach Scale, in the right frontotemporal V1 trigeminal region.
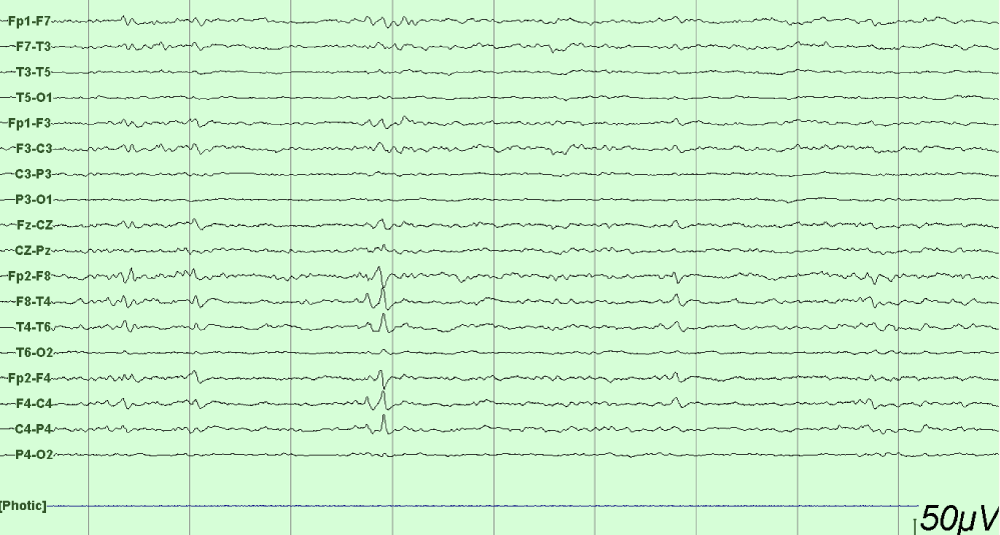
Figure 2: EEG longitudinal bipolar montage under anesthesia with epileptiform activity over the right frontotemporal region and continued generalized slowing.
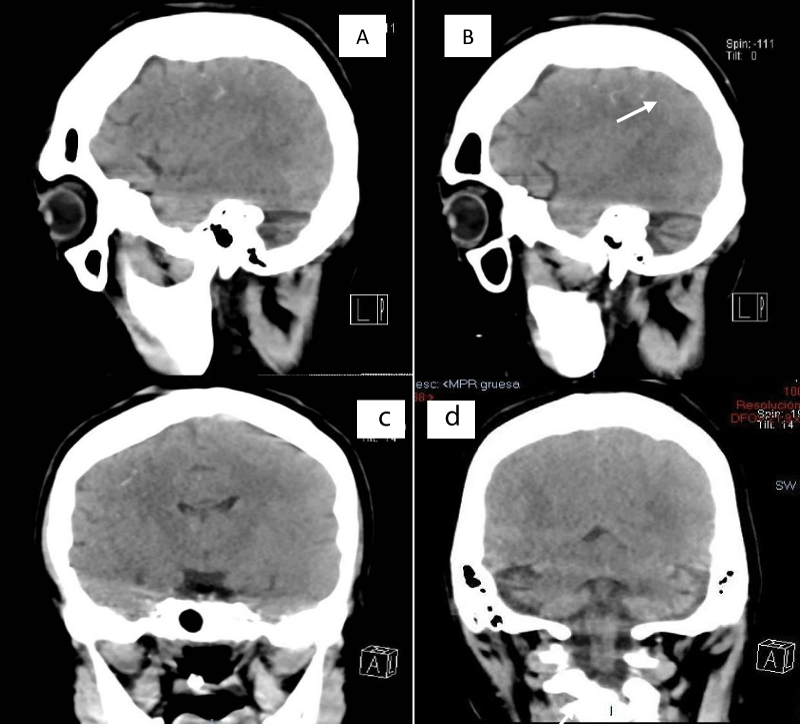
Figure 3: SWS brain involvement - Brain CT, non-contrast, sagittal, and axial showing calcifications in the right frontal and parietal regions and right cerebellar atrophy.
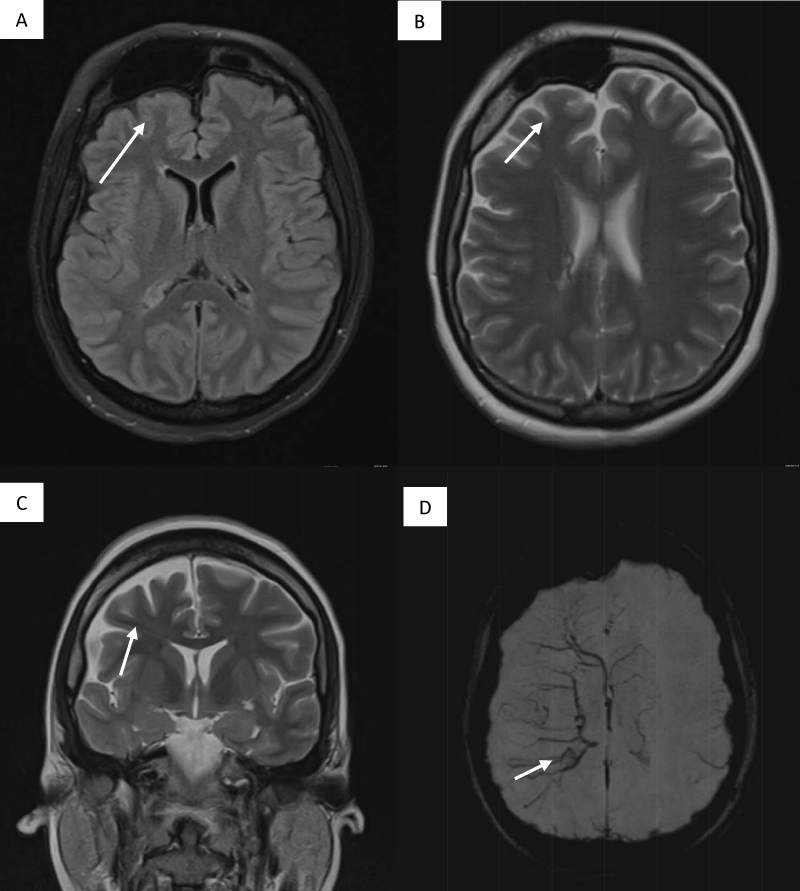
Figure 4: Brain MRI (A) FLAIR right cerebral atrophy and secondary frontal hyperostosis (B, C). T2 Asymmetric right hemisphere (D) SWI sequence abnormal capillary vessel over the right cerebral convexity.
Medical treatment is adjusted with levetiracetam 1500 mg BID, valproate acid 600-400-600 mg, topiramate 50 mg BID, clobazam 250 mg every 24 hours, and aspirin 100 mg every 24 hours, with clinical improvement. Currently, the patient has an improvement in the seizure rate, without presenting SE, currently with 3 seizures per month. She is following up with ophthalmology, physical rehabilitation, and neurology.
This case presentation highlights essential considerations for health professionals regarding the study of patients with this disease. SWS is a rare congenital vascular disorder characterized by a cutaneous capillary malformation, [11] that does not present mendelian inheritance. F. Parkes Weber whose 1922 paper led to the naming of the syndrome as Sturge-Weber. Historically SWS was categorized as one of the phakomatoses [12]. Characteristically, these patients present a capillary malformation, called PWB, a leptomeningeal malformation that typically enhances contrast on MRI [13]. Steve Roach in 1992 proposed a classification for the spectrum of “encephalotrigeminal angiomatosis”. The Roach Scale was used for SWS classification. Type I patients have both facial and leptomeningeal angiomas. Type II patients have isolated facial angioma (no CNS involvement). Type III patients have isolated leptomeningeal angioma [14]. Nonetheless, this outdated classification incorrectly implies that all patients with facial capillary malformation (without brain or eye involvement) have type II SWS. At present, we now know that the syndromic association to SWS and the risk of both brain and eye involvement can be more accurately stratified based on the extension and location of the facial capillary malformation [10]. SWS can also be accompanied by ocular vascular malformation, which can cause glaucoma, which has been reported as the most frequent ocular manifestation, with an incidence of 30% - 70%. [15] Pressure is treated with eyedrops, such as timolol and latanoprost, which decrease fluid production in the eye. Frequently, medical management fails, or the glaucoma is fulminant (particularly in infants) and surgery is required with shunt placement or other means of relieving ophthalmic pressure [3].
Patients with this diagnosis may present episodes of stroke-like events characterized by focal seizures, hemiparesis, speech, and cognitive disorders. Recurrent thrombosis has also been postulated as a potential cause. Based on these suppositions, preventative treatment using low-dose aspirin at 3 to 5 mg/kg/day has been proposed and used by many to limit or prevent stroke-like episodes and improve neurodevelopmental outcomes. [3,6,10]. This dual positive effect may reflect the prevention of thrombosis in the setting of venous stasis, thereby improving blood flow to the brain, and reducing the risk of seizures [10]. EEG can be normal or show asymmetrical background activity amplitude or frequency as well as interictal activities or seizures [1]. EEG is an important test to perform for SWS patients with a febrile illness and altered mental status (unexplained confusion or unresponsiveness), as they are at risk [6].
Some differential diagnoses are Blue rubber bleb nevus syndrome, Klippel-Trenaunay-Weber syndrome, PHACES (posterior fossa abnormalities, hemangiomas, arterial anomalies, cardiac, eye, and sternal anomalies), and Wyburg-Mason syndrome [1].
In the case of our patient, who has been diagnosed with SWS since birth, presenting the characteristic facial angioma in the right frontotemporal V1 trigeminal region, Type I according to the Roach Scale [14], who started with seizures at 4 months of age, which have been refractory according to the ILAE criteria [16]. The patient has a history of breakthrough seizures, generally triggered by upper and lower airway infections. Among the main causes of SE are the reduction of ASM at 34%, metabolic causes at 15%, and systemic infections at 7%. [1,17]. It has been observed that up to 57% of patients with SWS present SE during their evolution [18]. Among the challenges to be dealt with in this patient is proper management and avoiding triggering events that may exacerbate seizures.
This case illustrates the natural history of SWS, ranging from facial angioma to the onset of seizures before the first year of age, together with cognitive alterations and difficult-to-control seizures. The drugs most used in this disease are ASM and aspirin, which are postulated to reduce stroke events. Currently, the use of cannabidiol has also been reported for patients with refractory seizures, with which there is a decrease in seizures, improvement in quality of life, and subjective improvement in language, motor, and cognitive skills. The use of mTOR inhibitors has also been mentioned, hypothesizing a decrease in the risk of cerebrovascular events, normalizing the function of the affected vessels, with reports of significant improvement in anger control, depression, and quality of life. This disease presents with a wide spectrum of multisystemic manifestations, so a multidisciplinary approach is required to achieve the expected quality of life, despite the challenges that this disease provides us.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
- Singh AK, Keenaghan M. Sturge-Weber Syndrome. StatPearls. 2023. https://www.ncbi.nlm.nih.gov/books/NBK459163/
- Zeka N, Zeka B, Gerguri A, Bejiqi R, Retkoceri R, Maloku A, Zogaj L. Sturge-Weber syndrome and variability of clinical presentation. Med J Malaysia. 2023 Mar;78(2):145-148. PMID: 36988522.
- Comi A. Current Therapeutic Options in Sturge-Weber Syndrome. Semin Pediatr Neurol. 2015 Dec;22(4):295-301. doi: 10.1016/j.spen.2015.10.005. Epub 2015 Nov 11. PMID: 26706016; PMCID: PMC4943027.
- Comi AM, Sahin M, Hammill A, Kaplan EH, Juhász C, North P, Ball KL, Levin AV, Cohen B, Morris J, Lo W, Roach ES; 2015 Sturge-Weber Syndrome Research Workshop. Leveraging a Sturge-Weber Gene Discovery: An Agenda for Future Research. Pediatr Neurol. 2016 May;58:12-24. doi: 10.1016/j.pediatrneurol.2015.11.009. Epub 2016 Mar 16. PMID: 27268758; PMCID: PMC5509161.
- Vellieux G, Frazzini V, Pichit P, Dupont S, Gourfinkel-An I, Navarro V. A review of the natural history of Sturge-Weber syndrome through adulthood. J Neurol. 2022 Sep;269(9):4872-4883. doi: 10.1007/s00415-022-11132-9. Epub 2022 May 5. PMID: 35508811.
- De la Torre AJ, Luat AF, Juhász C, Ho ML, Argersinger DP, Cavuoto KM, Enriquez-Algeciras M, Tikkanen S, North P, Burkhart CN, Chugani HT, Ball KL, Pinto AL, Loeb JA. A Multidisciplinary Consensus for Clinical Care and Research Needs for Sturge-Weber Syndrome. Pediatr Neurol. 2018 Jul;84:11-20. doi: 10.1016/j.pediatrneurol.2018.04.005. Epub 2018 Apr 18. PMID: 29803545; PMCID: PMC6317878.
- Lo W, Marchuk DA, Ball KL, Juhász C, Jordan LC, Ewen JB, Comi A; Brain Vascular Malformation Consortium National Sturge-Weber Syndrome Workgroup. Updates and future horizons on the understanding, diagnosis, and treatment of Sturge-Weber syndrome brain involvement. Dev Med Child Neurol. 2012 Mar;54(3):214-23. doi: 10.1111/j.1469-8749.2011.04169.x. Epub 2011 Dec 23. Erratum in: Dev Med Child Neurol. 2012 Oct;54(10):957. multiple investigator names added. PMID: 22191476; PMCID: PMC3805257.
- Luat, A. F., Juhász, C., Loeb, J. A., Chugani, H. T., Falchek, S. J., Jain, B., … Pinto, A. (2019). Neurological Complications of Sturge-Weber Syndrome: Current Status and Unmet Needs. Pediatric Neurology, 98, 31–38. https://doi.org/10.1016/j.pediatrneurol.2019.05.013
- Yeom S, Comi AM. Updates on Sturge-Weber Syndrome. Stroke. 2022 Dec;53(12):3769-3779. doi: 10.1161/STROKEAHA.122.038585. Epub 2022 Oct 20. PMID: 36263782.
- Sánchez-Espino LF, Ivars M, Antoñanzas J, Baselga E. Sturge-Weber Syndrome: A Review of Pathophysiology, Genetics, Clinical Features, and Current Management Approache. Appl Clin Genet. 2023 Apr 24;16:63-81. doi: 10.2147/TACG.S363685. PMID: 37124240; PMCID: PMC10145477.
- Desai S, Glasier C. Sturge–Weber Syndrome. 2017. 377(9): e11. https://doi.org/10.1056/NEJMICM1700538
- Comi AM, McCann M, Singh P, Pevsner J. Sturge-Weber syndrome. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease. 2020; 2: 213–223. https://doi.org/10.1016/B978-0-12-813866-3.00014-X
- Pinto AL, Chen L, Friedman R, Grant PE, Poduri A, Takeoka M, Prabhu SP, Sahin M. Sturge-Weber Syndrome: Brain Magnetic Resonance Imaging and Neuropathology Findings. Pediatr Neurol. 2016 May;58:25-30. doi: 10.1016/j.pediatrneurol.2015.11.005. Epub 2015 Nov 26. PMID: 26706049.
- Roach ES. Neurocutaneous syndromes. Pediatr Clin North Am. 1992 Aug;39(4):591-620. doi: 10.1016/s0031-3955(16)38367-5. PMID: 1635798.
- Yan H, Hu M, Cui Y, Li L, Liang T. Clinical characteristics of infants with port-wine stain and glaucoma secondary to Sturge-Weber Syndrome. BMC Ophthalmol. 2022 Jun 9;22(1):260. doi: 10.1186/s12886-022-02476-x. PMID: 35681114; PMCID: PMC9185922.
- Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, Moshé SL, Perucca E, Wiebe S, French J. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010 Jun;51(6):1069-77. doi: 10.1111/j.1528-1167.2009.02397.x. Epub 2009 Nov 3. Erratum in: Epilepsia. 2010 Sep;51(9):1922. PMID: 19889013.
- Betjemann JP, Lowenstein DH. Status epilepticus in adults. Lancet Neurol. 2015 Jun;14(6):615-24. doi: 10.1016/S1474-4422(15)00042-3. Epub 2015 Apr 20. PMID: 25908090.
- Powell S, Fosi T, Sloneem J, Hawkins C, Richardson H, Aylett S. Neurological presentations and cognitive outcome in Sturge-Weber syndrome. Eur J Paediatr Neurol. 2021 Sep;34:21-32. doi: 10.1016/j.ejpn.2021.07.005. Epub 2021 Jul 9. PMID: 34293629.