
More Information
Submitted: February 19, 2021 | Approved: March 22, 2021 | Published: March 23, 2021
How to cite this article: do Amaral LC. Cortical spreading depolarizations in the context of subarachnoid hemorrhage and the role of ketamine. J Neurosci Neurol Disord. 2021; 5: 016-021.
DOI: 10.29328/journal.jnnd.1001045
Copyright License: © 2021 do Amaral LC. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Ketamine; Subarachnoid haemorrhage; Cerebral ischemia; Cortical spreading depolarizations
Keywords: ACA: Anterior Cerebral Artery; ADC: Apparent Diffusion Coefficient; ASAH: Aneurysmal Subarachnoid Haemorrhage; CBF: Cerebral Blood Flow; CSDs: Cortical Spreading Depolarizations; DCI: Delayed Cerebral Ischemia; ICP: Intracranial Pressure; MCA: Middle Cerebral Artery; MR: Magnetic Resonance; NMDA: N-Methyl D-Aspartate; rCBV: relative Cerebral Blood Volume; SAH: Subarachnoid Haemorrhage

Cortical spreading depolarizations in the context of subarachnoid hemorrhage and the role of ketamine
Leandro Custódio do Amaral*
Neurosurgeon, Rede Mater Dei de Saúde, Hospital Metropolitano Odilon Behrens and Fundação Benjamin Guimarães/Hospital da Baleia, Brazil
*Address for Correspondence: Leandro Custódio do Amaral, Rua Cristina, nº 452, apt 1302, Bairro Sion – Belo Horizonte Minas Gerais, Brazil, CEP 30.310-800; Tel: (+05531) 2552-0948/99685-0948; Email: [email protected]; [email protected]
Delayed cerebral ischemia (DCI) is one of the main complications of spontaneous subarachnoid haemorrhage and one of its causes is the cortical spreading depolarizations (CSDs). Cortical spreading depolarizations are waves of neuronal and glial depolarizations in which there is loss of neuronal ionic homeostasis with potassium efflux and sodium and calcium influx. In damaged brain areas and brain areas at risk, such as those adjacent to subarachnoid haemorrhage (SAH), CSDs induce microvascular vasoconstriction and, therefore, hypoperfusion and spread of ischemia. Several studies have been devoted to minimize secondary injuries that occur hours to days after an acute insult. Ketamine, a drug until recently contraindicated in the neurosurgical population for potentially causing intracranial hypertension, has re-emerged as a potential neuroprotective agent due to its pharmacodynamic effects at the cellular level. These effects include anti-inflammatory mechanisms, and those of microthrombosis and cell apoptosis controls, and of modulation of brain excitotoxicity and CSDs. A literature review was performed at PubMed covering the period from 2002 to 2019. Retrospective studies confirmed the effects of ketamine on the control of CSDs and, consequently, of DCI in patients with SAH, but did not show improvement in clinical outcome. The influence of ketamine on the occurrence/development of DCI needs to be further confirmed in prospective randomized studies.
Delayed cerebral ischemia (DCI) is one of the main complications of spontaneous aneurysmal subarachnoid haemorrhage (aSAH), being responsible for the occurrence of cerebral infarctions, which determine poor clinical outcomes. The incidence of DCI is up to 40% and has a multifactorial origin, including cerebral vasospasm, neuroinflammation, microthromboses and cortical spreading depolarizations (CSDs) [1].
CSDs are waves of almost complete depolarization of both the neuronal and glial cells [2]. In damaged brain areas and in brain areas at risk, such as ischemic penumbra zones and those zones adjacent to aSAH, CSDs induce microvascular vasoconstriction and, therefore, hypoperfusion and ischemia spreading. That is, under pathological circumstances, CSDs give rise to oxidative stress, exacerbate hypoxia, and cause neuronal death [3,4].
Within this context of brain injury, a large number of researches have been devoted to minimize secondary injuries, which occur hours to days after the acute insult [5]. Thus, control of CSDs has gained increasing importance, but remains largely unknown if they may be pharmacologically modulated in the human brain [4]. Ketamine, a drug until recently contraindicated in the neurosurgical population for potentially causing intracranial hypertension, has re-emerged as a potential neuroprotective agent due to its pharmacodynamic effects at the cellular level [5].
Delayed cerebral ischemia (DCI) is one of the main complications of spontaneous aneurysmal subarachnoid haemorrhage (aSAH), being responsible for the occurrence of cerebral infarctions, which determine poor clinical outcome [1]. The incidence of DCI is up to 40% [1,6]. Nevertheless, the incidence of infarctions verified by cranial computed tomography (CT) is only 10% - 13% [1]. This is because the presence of small parenchymal and cortical lesions is underestimated due to the technical limitations of cranial CT when compared to magnetic resonance (MR) especially when using diffusion images [6].
Traditionally, DCI have been attributed to the presence of proximal vasospasm and great effort has been made in basic and clinical research to prevent angiographic vasospasm [7]. However, DCI may also occur in the absence of angiographic vasospasm [6,7]. Thus, the hypothesis that cerebral vasospasms are the only cause of DCI has become controversial. In the anatomopathological literature, there is a predominance of cortical infarcts over territorial infarcts, as well as heterogeneous perfusion patterns, varying from hypoperfusion to hyperfusion, found in positron emission tomographies of patients with aSAH and DCI have also suggested the presence of other mechanisms contributing to the occurrence of DCI [7]. In addition, the positive predictive value of the presence of vasospasms for the occurrence of DCI is low. Finally, the use of Clazosentan, an endothelin A receptor antagonist, reduced the relative risk of angiographic vasospasm in 65%, but it did not significantly improve the clinical outcome [7]. All of these facts reaffirm the multifactorial origin of DCI, including neuroinflammation, microthromboses and CSDs in addition to cerebral vasospasm [1].
CSDs are waves of almost complete depolarization both neuronal and glial ones [2-4]. They are measured by the glutamate release into the extracellular space with consequent activation of glutamatergic ionotropic receptors, potassium (K+) efflux, sodium (Na+) and calcium (Ca2+) influx, overcoming the effects of adenosine triphosphate-dependent sodium-potassium pumps and leading to a complete disruption of ionic gradients [4]. Loss of neuronal ionic homeostasis leads to cellular oedema, dendritic distortions, and depression of neuronal activity [3,4].
CSDs may occur spontaneously after mechanical injury, hypoxia, ischemia, hypoglycaemia, high extracellular concen-trations of K+ and nitric oxide reductions by haemoglobin degradation products. All of these pathological conditions frequently occur after aSAH [3].
Normal and injured brains have different responses to CSDs. In the normal brain, CSDs induce arteriolar dilation and elicit a wave of hyperaemia which supplies tissues with enough energy to re-establish ionic balance [3,4,8]. In these situations, cerebral blood flow increases over 100% [3]. In tissues at risk, such as areas adjacent to aSAH, an inverse hemodynamic response with microvascular constriction and hypoperfusion may be seen [3,4,6,8]. Under pathological circumstances, CSDs may produce oxidative stress, worsen hypoxia and induce neuronal death [3,4].
Despite studies animals dating from 1944, the electrophysiological confirmation of CSDs in the human brain and its pathophysiological role in severe neurological conditions had not been established until the last decade [3]. Currently, there is enough evidence showing the significant role of CSDs in migraine with aura, transient global amnesia, ischemic strokes, intraparenchymal haemorrhages, cranioencephalic trauma and Asah [3,8].
The incidence of CSDs in patients is very high, with rates ranging from 66% to 100%, and their presence is associated with the occurrence of DCI in 44% to 88% of cases [3].
CSDs in the cerebral cortex are involved in the pathogenesis both of acute ischemia and delayed cerebral ischemia after aSAH [9,10]. In an observational multicenter study of 54 patients electrocorticographically monitored using subdural electrodes, early focal brain injury after aSAH was associated with early CSDs. Thirty-three of 37 patients with early focal brain injury (intracerebral hemorrhage and/or hypodensity) in contrast to 7 of 17 without displayed CSDs during days 1-4 (p < 0,001) [9].
Aneurysmal subarachnoid haemorrhage may induce several ischemic patterns in imaging exams. They include territorial infarcts, lacunar infarcts in white matter and focal or diffuse cortical lesions. The first two ones develop as a consequence of moderate or severe, proximal and/or distal vasospasm visible on angiography [6]. On the other hand, laminar and focal cortical ischemic lesions caused by CSDs are different from those caused by elevations of intracranial pressure or hypoxia, when there is a diffuse cortical involvement [6,11]. Cortical lesions of CSDs occur in areas covered by haematomas at the depth of cerebral sulci, since haemoglobin degradation products are responsible for causing severe acute vasoconstriction in the cortical microcirculation [6,11]. The magnetic resonance (MR) study in patients with acute aSAH has shown symmetrical lesions in multiple vascular territories, especially in the most medial territories of middle cerebral artery (MCA) and anterior cerebral artery (ACA) [10].
With regard to the MR findings, they provide powerful tools to detect and assess co-occurring cerebral hemodynamic and cellular changes during CSDs [12]. Gradient echo sequences are sensitive to changes in cerebral blood flow (CBF) and metabolism that occur during the CSDs wave propagation, whereas diffusion-weighted imaging is capable of detecting water shifts from extracellular to intracellular compartments that are associated with depolarization and give rise to dramatic cellular swelling, which is characteristic of CSDs [13]. Transient apparent diffusion coefficient (ADC) declines appear simultaneously with a negative direct current potential shift (Figure 1) [13]. Transient regional hyperperfusion follows ADC decreases thus supporting the observation of increased energy demand from repolarization [13]. A prolonged period of hypoperfusion follows the initial relative cerebral blood volume (rCBV) (Figure 2). Transient ADC and rCBV changes were limited to the cortex [13].
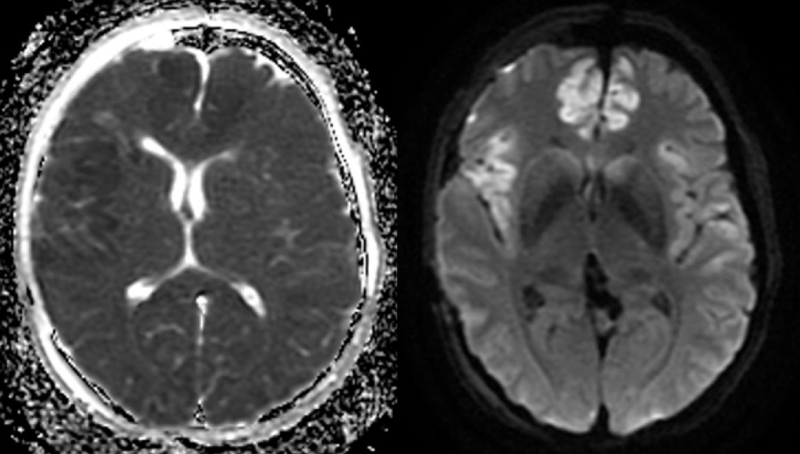
Figure 1: Cortical ADC decline (left) and diffusion hypersignal (right) on the right insular and anterior interhemispheric regions on day 4 after an aSAH.
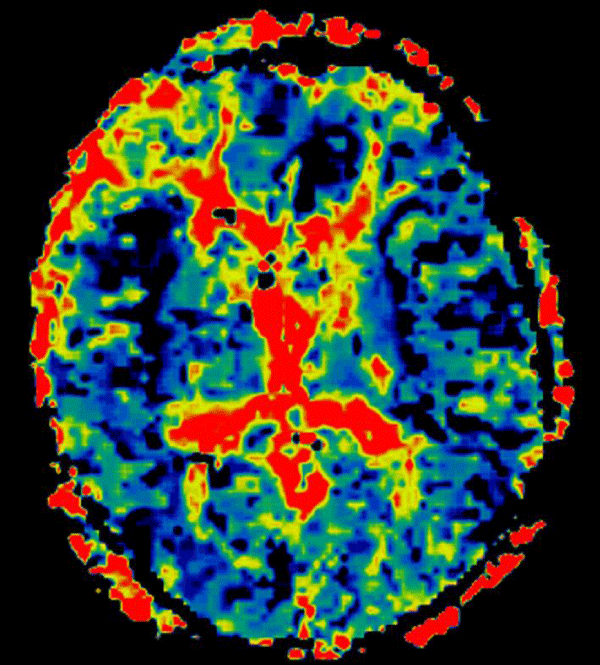
Figure 2: Cortical hypoperfusion detected on the right insular and anterior interhemispheric regions on day 4 after an aSAH.
CSDs are associated to functional neurological damage, neuronal necrosis, neurological deterioration, and poor clinical outcomes [2,3]. Therefore, the control of CSDs has a growing importance [3]. But, the possibility of a pharmacological therapy to modulate CSDs in humans still remains unknown [4]. However, preliminary studies have suggested that high doses of analgesics and sedatives may influence the occurrence of CSDs [1-5]. Within this context, n-methyl D-aspartate (NMDA) receptors blockers, such as Ketamine, has shown promising results due to their potential neuroprotective effects [1,4,6]. Until recently, the use of ketamine was contraindicated in the neurosurgical population for potentially causing intracranial hypertension [5]. However, this is not the reality, especially when regarding intubated patients whose normocarbia is maintained [4,5].
Neuroprotective effects of ketamine and potential reduction in volumes both of haemorrhagic necrosis and of infarcted areas in focal ischemia result from multiple processes. These effects include not only anti-inflammatory mechanisms and those of microthrombosis and cell apoptosis controls, but also of modulation of brain excitotoxicity and of CSDs [5].
The first and most obvious process attenuated by ketamine is excitotoxicity, a dysregulation of neuronal calcium homeostasis resulting in excessive glutamate binding to postsynaptic NMDA receptors [5]. After primary brain insult, the low supply of oxygen and glucose compromises the ATP-dependent Na+/K + pump, leading to an intracellular accumulation of Na+ and an extracellular accumulation of K+. This leads to loss of the Na+ electrogenic gradient and consequent interruption of the Na+/Ca2+ exchange that for not releasing Ca2+ into the extracellular space, allows its own intracellular accumulation. Nevertheless, Ca2+ also enters the cells through the activation of voltage sensitive Ca2+ channels. Elevation of intracellular Ca2+ induces uncontrolled fusion of vesicles containing glutamate to the presynaptic membrane, determining glutamate exocytosis and increased postsynaptic Ca2+ [5]. Accumulation of Ca2+ in the intracellular space induces increased DNA fragmentation, degradation of the neuronal cytoskeleton and apoptosis5. Extrasynaptic NMDA receptors may induce cell death by a distinct mechanism, by directly binding with proteins involved in cell death such as neuronal nitric oxide synthase [5]. In addition, extrasynaptic NMDA receptors are related to flow through glutamate receptors to cell surface favouring the cell hyperexcitability and vulnerability to a secondary injury [5].
Ketamine may mitigate the excitotoxicity through non-competitive antagonism of NMDA receptors, reducing calcium influx [5]. Ketamine also attenuates the release of glutamate interfering with the fusion of vesicles containing glutamate to the presynaptic membrane [5]. Thus, ketamine slows down the excitatory transmission and antagonises excessive glutamate release [5]. Most recent studies have shown ketamine also have an intense extrasynaptic effect, inhibiting the selective activation of neurotoxic NMDA receptors [5]. Antiglutamatergic effects of ketamine may play an important role in the treatment of DCI after aSAH and vasospasm since these conditions are associated with elevations of extracellular glutamate that, in its turn, is related to the clinical evolution and to the emergence of neurological symptoms [5].
In addition to the control of excitotoxicity, studies in models of focal ischemia have shown that ketamine is able to reduce cell apoptosis through a hyper-regulation of B2-cell lymphoma expression, a protein with an anti-apoptotic effect. Thus, ketamine prevents neuronal and glial apoptosis [5]. Studies conducted in the 1990s, also showed that neuroprotective effects of ketamine occur, partly, by the inhibition of catecholamine release. This occurs because they are able to induce neuronal apoptosis and their metabolites have direct toxic effects [5]. Thus, the decreased catecholamine release would benefit cell survival.
Another important property of ketamine, both for its analgesic effect and for its neuroprotective action, is its anti-inflammatory capacity [5]. Ketamine mediates the inhibition of the production of tumour necrosis factor (TNF-α) and interleukins 6 and 8, thus mitigating the neuroinflammatory response to hypoxia [5]. Another new anti-inflammatory mechanism of ketamine is to inhibit the activation of endothelial cells by HMGB1, a protein whose high levels predict non survival and increased neuroinflammation in models of aSAH [5]
Microthromboses are deleterious processes, which also contribute to neuronal damage after aSAH, since they impair oxygen and glucose distribution to neurons, determining local hypoxia [5]. Microthromboses may mediate DCI in patients with aSAH. Another important consequence of microthromboses is an intense glutamate release from platelets during their own aggregation, thus favouring excitotoxicity [5]. Currently, microthromboses are known to play a role as mediators of DCI after aSAH in association with cerebral vasospasm cerebral [5]. Ketamine plays an additional role in the control of DCI through its antiplatelet effect, which determines reductions in the accumulation of intracellular Ca2+ and formation of thromboxane A2 and, finally, consequent reductions in platelet aggregation and formation of microthrombosis 5].
Ketamine has several other properties which are helpful in neurointensivism and in the treatment of patients with aSAH. It provides protection against excitotoxicity, and secondary damages to status epilepticus, but as previously mentioned above, it is also able to suppress CSDs [3,5].
Ketamine seems to be useful in the treatment of status epilepticus for having, during prolonged epileptiform activity, a suppressed activity of GABAergic receptors and hyperstimulation of postsynaptic NMDA receptors [5]. Thus, due to the decreased GABAergic activity and increased glutamatergic activity in prolonged epileptiform activities, ketamine has shown to be effective in the control of status epilepticus when GABAergic anticonvulsants have failed [5]. A systematic review with 110 patients with status epilepticus has shown their response rate to ketamine was 56.5%. Therefore, ketamine provides a significant neuroprotection against neuronal death after status epilepticus [5].
From the hemodynamic point of view, ketamine also play an important role in neurointensivism. Ketamine helps to maintain hemodynamic stability and improves the cerebral blood flow, reducing the need for vasopressors to maintain the cerebral perfusion pressure [1-4].
In view of all possible beneficial effects of ketamine and its effects on the control of CSDs associated to evidence that these are not related to DCI, clinical studies in humans were started to confirm these facts.
Hertle, et al. [2] tested the influence of several analgesics and sedatives such as midazolam, propofol, fentanyl and ketamine in the occurrence of CSDs in 115 patients with several diagnoses, such as ASAH, traumatic brain injuries and ischemic strokes and intraparenchymal haemorrhagic strokes [2]. In this study, only the administration of ketamine was associated to the reduction in the occurrence of CSDs [2-4].
Dreier, et al. [11] have already shown ketamine’s ability to suppress CSDs in patients with aSAH with a dose of 2-3 mg/kg/h [14]. Nevertheless, data were still conflicting since a case in which CSDs had not been suppressed after a dose of ketamine (2 mg/kg/h) had already been reported [15]. These findings suggested a ketamine dose-dependent effect which was later shown by Hertle, et al. [2]. There is an inverse linear association between doses of ketamine and the presence of CSDs. Ketamine was able to suppress both isolated CSDs and clustered ones [2].
Propofol was also able to suppress the occurrence of clustered CSDs, but to a lesser extent than ketamine [2]. In addition, the use of propofol as an adjuvant to increase the level of sedation resulted in increased doses of vasopressors in up to 80% of cases [6]. On the other hand, midazolam has shown a positive association with the occurrence of clustered CSDs, increasing their frequency [2]. Midazolam leads to a reduction in the energy use by the brain, a metabolic depression mediated by the GABAergic activity which aggravates the failure of energy-dependent ionic pumps and, therefore, increases the generation and spreading of CSDs [2].
In 2016, Von der brelie, et al. [1] published a study that aimed to evaluate the safety and influence of ketamine on occurrence of cerebral infarctions associated to DCI in patients with aSAH. In this study, a group of patients sedated with opidoids (fentanyl or sulfentanil) and benzodiazepines (midazolam) was compared to another in which ketamine with doses of up to 500 mg/hour had been associated to the basic regimen to reach the appropriate sedation level or to control the intracranial hypertension [1]. The occurrence of cerebral infarctions associated to DCI was significantly lower in the group using ketamine (7.3%) than that in the group without ketamine (25%); p = 0.046 [1]. It is hypothesized that this had occurred influenced by ketamine in the occurrence of CSDs and DCI in patients with aSAH, but this study did not include electrophysiological monitoring and the effects of ketamine in this context are still not clear and require further studies [1]. The use of ketamine allowed the normalization of the intracranial pressure (ICP) and appropriate sedation at 92.7%, with no reports of critical increases of ICP [1]. Actually, ketamine reduced the ICP in 93.1% of patients [1]. Therefore, the use of ketamine is useful and safe as for ICP, since the possible increase of ICP due to the increased cerebral blood flow does not routinely occur in sedated and ventilated patients thanks to the decreased cerebral metabolism by other sedatives [1]. Through its desirable effects in increased systemic and pulmonary pressure and cardiac output, ketamine allowed a significant decrease in the dose of vasopressors in 56.3% of patients [1]. This fact prevents the negative effect of vasopressor on the perfusion of organs and extremities, as well as the development of pulmonary oedema [1]. Despite all these findings, there were no significant differences in complication rates (pneumonia, venous thromboembolism and pressure ulcers) between groups with and without ketamine [1], except for gastroparesis, which was more frequent in the group using ketamine, 87.8% versus 50%; p = 0.008 [1]. Moreover, patients with ketamine were maintained sedated longer, 21.2 days versus 13.5 in the group without ketamine, p = 0.001 [1]. There were no statistically significant differences in the clinical outcome and mortality between the group who used and who did not use ketamine [1]. Nevertheless, patients allocated to the group using ketamine were more severe, WFNS (World Federation of Neurosurgical Societies grading system) grade 3 versus WFNS grade 2 in the group without ketamine, p = 0.02 [1]. However, this was a retrospective study, and it did not have electrophysiological data showing the influence of ketamine in CSDs and did not temporally relate the administration of ketamine with the occurrence of DCI.
In 2019 Santos, et al. published the largest study on the effect of ketamine on CSDs in aSAH patients. This retrospective cohort of 66 patients electrocorticographically monitored in a neurointensive care unit with a single subdural electrode strip found that ketamine administration was associated with a reducer CSDs incidence with a small and clinically irrelevant increase in ICP. The blood pressure also increased. However, in the DCI phase, this is rather a positive side effect. Importantly, CSDs were most potently inhibited by doses of ketamine that are above clinically recommended doses, but were nonetheless given by neurointensivists in individual cases in which there was a need of stronger sedation. The retrospective study design led to the selection bias that the sicker patients were treated with ketamine. Thus, patients receiving ketamine more frequently showed WFNS 4 or 5 values at admission and had more severe aSAH on the modified Fisher scale, which is the most likely reason why ketaminetreated patients had a worse outcome. However, it cannot be completely excluded that the ketamine group experienced more severe secondary complications due to, for example, stronger sedation [8].
In vitro and animal studies show a series of benefits of ketamine to the control of excitotoxicity; to the reductions of cell apoptosis, of neuroinflammation and of microthrombosis or to the suppression of CSDs and of associated DCI. Thus, ketamine, has gained importance due to its neuroprotective potential. Several studies have shown the effect of ketamine on the control of CSDs in patients with cranioencephalic traumas. Nevertheless, the literature referring to the role of ketamine in patients with aSAH is still scarce. Case reports strongly suggest the effect of ketamine on the control of DCI and therefore, of DCI in patients with aSAH. Retrospective studies confirmed these findings but did not show an improvement in the clinical outcome. The influence of ketamine in the occurrence/development of ICT still needs to be confirmed in randomized prospective studies [1].
Declarations
This is a review article and therefore ethical approval does not apply in this case.
Consent to participate: Informed consent to participate is not required in this type of study.
Authors’ contributions: The author was responsible for all material preparation, data collection, analysis, article writing and revision.
- Von der Brelie C, Seifert M, Rot S, Tittel A, Sanft C, et al. Sedation of Patients with Acute Aneurysmal Subarachnoid Hemorrhage with Ketamine Is Safe and Might Influence the Occurrence of Cerebral Infarctions Associated with Delayed Cerebral Ischemia. World Neurosurg. 2017; 97: 374-382. PubMed: https://pubmed.ncbi.nlm.nih.gov/27742511/
- Hertle DN, Dreier JP, Woitzik J, Hartings JA, Bullock R, et al. Effect of analgesics and sedatives on the occurrence of spreading depolarizations accompanying acute brain injury. Brain. 2012; 135: 2390-2398. PubMed: https://pubmed.ncbi.nlm.nih.gov/22719001/
- Sánchez-Porras R, Zheng Z, Sakowitz OW. Pharmacological modulation of spreading depolarizations. Acta Neurochir Suppl. 2015; 120: 153-157. PubMed: https://pubmed.ncbi.nlm.nih.gov/25366616/
- Welling L, Welling MS, Teixeira MJ, Figueiredo EG. Cortical spread depolarization and ketamine: a revival of an old drug or a new era of neuroprotective drugs? World Neurosurg. 2015; 83: 396-397. PubMed: https://pubmed.ncbi.nlm.nih.gov/25644895/
- Bell JD. In Vogue: Ketamine for Neuroprotection in Acute Neurologic Injury. Anesth Analg. 2017; 124: 1237-1243. PubMed: https://pubmed.ncbi.nlm.nih.gov/28079589/
- Weidauer S, Vatter H, Beck J, Raabe A, Lanfermann H, Seifert V, et al. Focal laminar cortical infarcts following aneurysmal subarachnoid haemorrhage. Neuroradiology. 2008; 50: 1-8. PubMed: https://pubmed.ncbi.nlm.nih.gov/17922121/
- Woitzik J, Dreier JP, Hecht N, Fiss I, Sandow N, et al. Delayed cerebral ischemia and spreading depolarization in absence of angiographic vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2012; 32: 203-212. PubMed: https://pubmed.ncbi.nlm.nih.gov/22146193/
- Santos E, Olivares-Rivera A, Major S, Sánchez-Porras R, Uhlmann L, et al. Lasting s-ketamine block of spreading depolarizations in subarachnoid hemorrhage: a retrospective cohort study. Crit Care. 2019; 23: 427. PubMed: https://pubmed.ncbi.nlm.nih.gov/31888772/
- Eriksen N, Rostrup E, Fabricius M, Scheel M, Major S, et al. Early focal brain injury after subarachnoid hemorrhage correlates with spreading depolarizations. Neurology. 2019; 92: e326-e341. PubMed: https://pubmed.ncbi.nlm.nih.gov/30593517/
- Wartenberg KE, Sheth SJ, Michael Schmidt J, Frontera JA, Rincon F, et al. Acute ischemic injury on diffusion-weighted magnetic resonance imaging after poor grade subarachnoid hemorrhage. Neurocrit Care. 2011; 14: 407-415. PubMed: https://pubmed.ncbi.nlm.nih.gov/21174171/
- Dreier JP, Sakowitz OW, Harder A, Zimmer C, Dirnagl U, et al. Focal laminar cortical MR signal abnormalities after subarachnoid hemorrhage. Ann Neurol. 2002; 52: 825-829. PubMed: https://pubmed.ncbi.nlm.nih.gov/12447937/
- Umesh Rudrapatna S, Hamming AM, Wermer MJ, van der Toorn A, Dijkhuizen RM. Measurement of distinctive features of cortical spreading depolarizations with different MRI contrasts. NMR Biomed. 2015; 28: 591-600. PubMed: https://pubmed.ncbi.nlm.nih.gov/25820404/
- de Crespigny A, Röther J, van Bruggen N, Beaulieu C, Moseley ME. Magnetic resonance imaging assessment of cerebral hemodynamics during spreading depression in rats. J Cereb Blood Flow Metab. 1998; 18: 1008-1017. PubMed: https://pubmed.ncbi.nlm.nih.gov/9740104/
- Sakowitz OW, Kiening KL, Krajewski KL, Sarrafzadeh AS, Fabricius M, et al. Preliminary evidence that ketamine inhibits spreading depolarizations in acute human brain injury. Stroke. 2009; 40: e519-522. PubMed: https://pubmed.ncbi.nlm.nih.gov/19520992/
- Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain. 2009; 132: 1866-1881. PubMed: https://pubmed.ncbi.nlm.nih.gov/19420089/