Research Article
Role of plants, environmental toxins and physical neurotoxicological factors in Amyotrophic lateral sclerosis, Alzheimer Disease and other Neurodegenerative Diseases

Mauro Luisetto1*, Naseer Almukhtar2, Ahmed Yesvi Rafa3, Behzad Nili Ahmadabadi4, Ghulam Rasool Mashori5, Farhan Ahmad Khan6, Ram Kumar Sahu7, Gamal Abdul Hamid8 and Khaled Edbey9
1Applied Pharmacologist, Independent Researcher, Italy
2Professor, Physiology, College of Medicine, University of Babylon, Hilla, Iraq
3Founder and President, Yugen Research Organization (YRO), Independent Researcher, Bangladesh
4Pharm D/PhD Innovative Pharmaceutical Product Development Specialist, USA
5Professor of Pharmacology, People University of Medical & Health Sciences for Women, Nawabshah, Pakistan
6Associate Professor and Head, Department of Pharmacology, Government Medical College, Shahdol, M.P, India
7Associate Professor, Pt Deendayal Upadhyay Memorial Health Science and Ayush University of Chhattisgarh, Raipur, India
8Professor, Hematology Oncology, University of Aden, Aden, Yemen
9Professor, Department of Chemistry, University of Benghazi, Libya
*Address for Correspondence: Mauro Luisetto, Applied Pharmacologist, Independent Researcher, Italy, Email: [email protected]
Dates: Submitted: 18 February 2019; Approved: 01 March 2019; Published: 04 March 2019
How to cite this article: Luisetto M, Almukhtar N, Rafa AY, Ahmadabadi BN, Mashori GR, et al. Role of plants, environmental toxins and physical neurotoxicological factors in Amyotrophic lateral sclerosis, Alzheimer Disease and other Neurodegenerative Diseases. J Neurosci Neurol Disord. 2019; 3: 001-086. DOI: 10.29328/journal.jnnd.1001019
Copyright License: © 2019 Luisetto M, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Als; Pd; Ad; Plants neurotoxins; Intracellular neuronal inclusion; Oxidative damages; Epidemiology; Sports; Pesticides; Cianobacteria; Electromagnetic field; Computational biology

Abstract
Aim of this work is to verify the effect of some neurotoxins, physical factors and geography in presentation of some Relevant Neurological disorder like some form of ASL, PD, AD.
The geographic diffusion of the ASL/PD in west pacific (GUAM foci), and mutation of SOD 1 and other mutations are interesting facts to verify the recent literature about the neurotoxic process.
Related to the references presented a global conclusion about the pathogenetic progression of some neurological disease will be produced as instrument for new hypothesis and for the introduction of new innovative therapeutic strategies.
Introduction
ASL is a progressive neuronal disease involving the motoneuron systems.
A Neurodegenerative process (anterior spinal cord and CNS: part of cortico-spinal tract) whit neuroinflamation (by astrocytes and microglia activation).
MOTORNEURON is interested: anterior horn spinal cord and bubble and piramidal way degeneration.
Intracellular inclusions (NOT AMYLOID) are common in some form: UBIQUITIN POSITIVE (prot. TDP-43, TAR DNA BINDING PROTEIN, FUS: FUSED IN SARCOMA, SOD1).
Misfolded proteins are associated with various neurodegenerative diseases:
Proteinopathy ------------------- Aggregating protein(s)
Alzheimer’s disease / Amyloid beta peptide; Tau
PD /α-synuclein
Huntington’s disease /Huntingtin with tandem glutamine repeats
Amyotrophic lateral sclerosis /Superoxide dismutase 1
Spongiform encephalopathies / Prion proteins
From literature MULTIFACTORIAL CAUSES are involved in ALS pathology and progression: unknown etiology for high number of cases, viruses and evnvironmental toxic substanties are not clearly proved.
SN is more sensible vs other tissue to ischemic damages because more high metabolism and oxigen need.
Ipoxia cause reduced ATP increase of intracellular CA++, increase of intracellular NA+ and water intake by the cell with final cell lisys.
After various damages Microglia and astrocite release proinflamatory citokine and Ros.
Reactive gliosys stimuly like trauma, ischemia, infectious event, cconvulsion, tox ic subtantie, CNS pathology and other.
Reactive gliosys is a defence reaction vs the damages stimuly: it produce fagocite activations and growth factor production for the repair activity. (TNF alfa, IL 1beta that activates the caspases and activated the apoptotic process).
The neurotrasmitters Glutammate show eccitotoxicity effects: if in high concentration after ischemic damages or or in example after some plants toxic agents like from latirus.
Glutammate in high concentration acts towards NMDA and AMPA receptor: open ca++ channels and caus se cell death.
After proteasys activation, phospholipases, cytoskeleton disorganization and ATP sintesys inhibition.
So Oxidative stress, genetic disorder, mitochondrial disfuntion, glutamate exitotoxicity, axon transport, proteosoma disfuncion whit protein aggregation, mRNA processing process disregulaions, neuroinflamation, neurotrofic factors deficit and other contribute in starting and promoting ALS.
Before analyzing factors related in some neuronal disease like ALS is interesting focus on brain and Nervous Tissue Metabolism and some patogenetic movens involved in neuronal toxicology.
Some peculiar characteristics are useful to set correctly the disease: in example GUAM foci of ALS /PD Geographic diffusion of some Genetic mutations (of SOD1 et other), and also involvement with some kind of sports (football) and the effect of physical environmental Factors and a cluster presentation of the cases.
In many situation the neuronal oxidative metabolism should be investigated effectively (Figures 1-6).
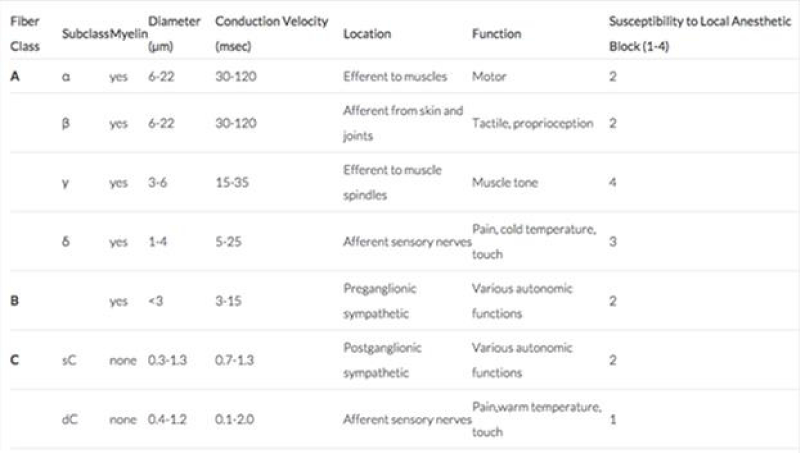
Figure 1: Diameter and other characteristic of neuronal fiber (from reference).
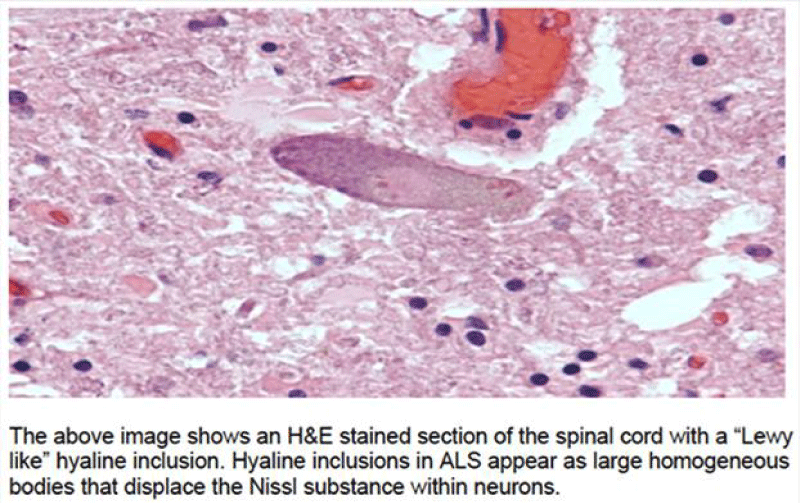
Figure 2: (From literature).
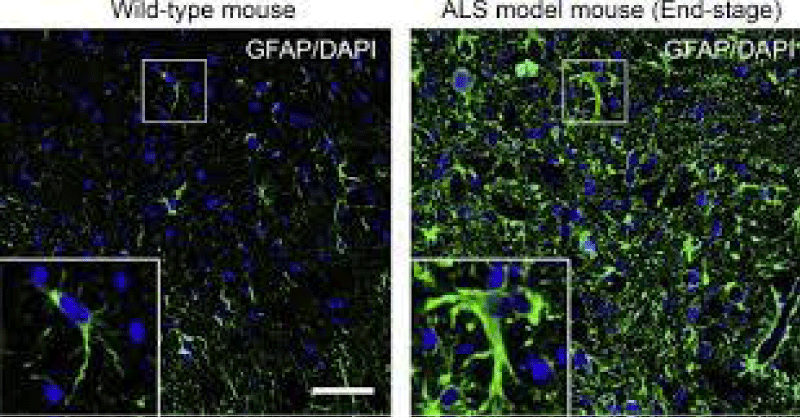
Figure 3: ALS MODEL MAUSE histology.
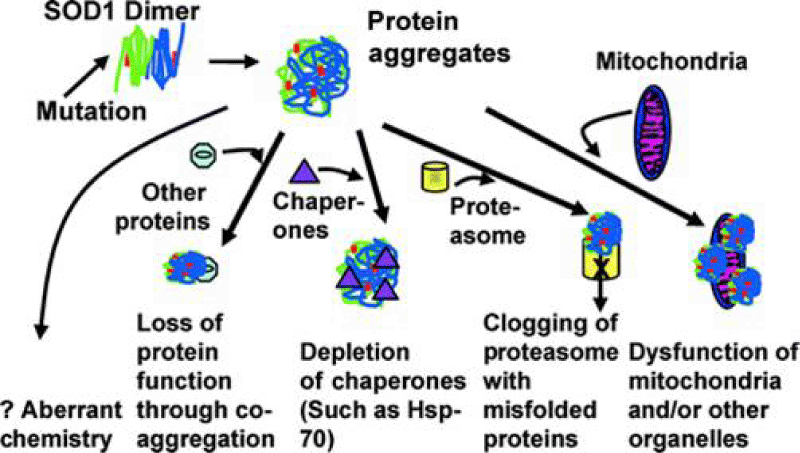
Figure 4: SOD 1 pathways (From lliterature).
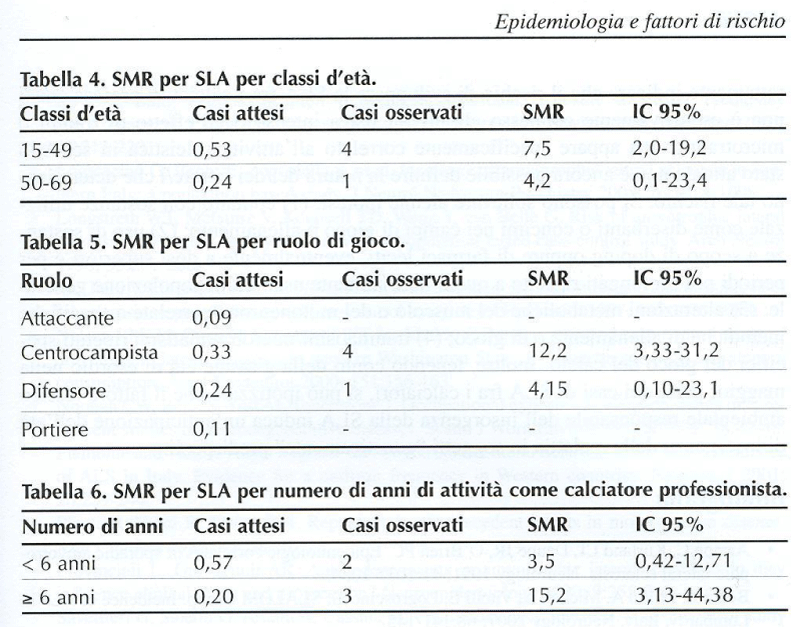
Figure 5: A. CHIO’ standardized mortality rate (From reference).
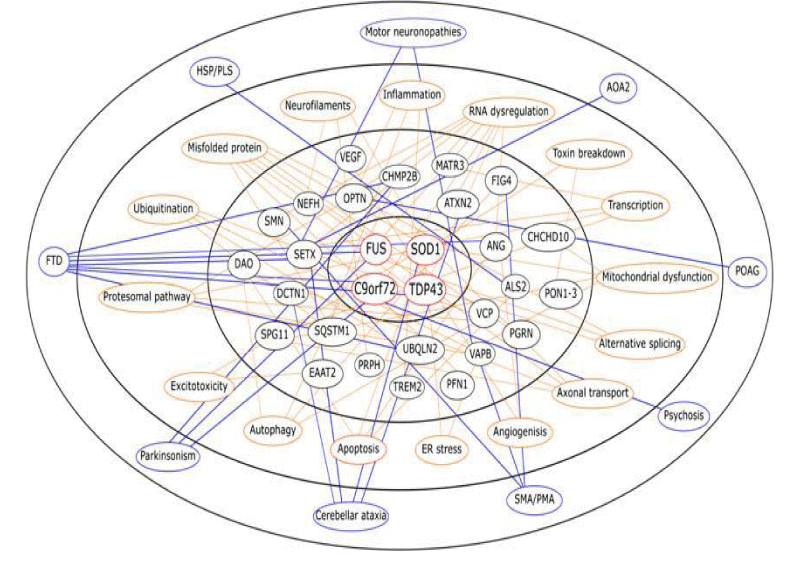
Figure 6: ALS mutation and pathogenesis from Pathogenesis of amyotrophic lateral sclerosis. Sarah Morgan and Richard W. Orrell 2016.
Material and Methods
With an observational approach some relevant literature was analyzed in order to produce a global hypotesis related to the pathological movens in some relevant neurotoxic disease.
All the literature where founded on PUBMED biomedical database.
After this review it will be presented and experimental project to verify this conclusion in an in vitro model about the related factors that contributes to the progression of disease and the one that can reduce this.
Results
From scientific literature
Kevin A Henry et al., “Geographic areas with higher localized incidence rates of ALS (e.g., geographic clustering) have been reported in specific regions world-wide. The most notable examples include a higher incidence of the Western Pacific form of ALS in the 1950s on the island of Guam [18], on the Kii Peninsula in Japan [19], and in southwestern New Guinea’’ [39].
“Motivated by investigations into potential environmental causes of ALS in the Western Pacific and other recent reports of localized clusters of ALS elsewhere in the world [21–30], researchers are increasingly using spatial analysis methods to map geographic variations in ALS incidence and to identify localized clusters of ALS [31]. The main goal of these investigations has been to locate geographic clusters of ALS and develop hypotheses about possible environmental risk factors responsible for the clusters” [31].
According Anna Falkowska et al., “Glycogen metabolism has important implications for the functioning of the brain, especially the cooperation between astrocytes and neurons [61].
In a glycogen deficiency (during hypoglycemia) glycogen supplies are used to generate lactate, which is then transported to neighboring neurons.
During periods of intense activity of the nervous system NS, when the energy demand exceeds supply, astrocyte glycogen is immediately converted to lactate, some of which is transported to the neurons.
Glycogen from astrocytes functions as a kind of protection against hypoglycemia, ensuring preservation of neuronal function.
This review goes on to emphasize that while neurons and the astrocytes differ in metabolic profile, they interact to form a common metabolic cooperation.”
Interactions between the astrocytes and neurons have the purpose of not only meeting the energy needs of these cells, but are also important in the control of many important brain functions as homeostasis of the body or memory consolidation. Metabolic disorders in the interactions between neurons and astrocytes can cause neuro-degenerative disorders and a number of other pathologies’’.
Anna Dawson, “The mass of the brain is about only 2% of the global body mass, yet its energy requirement is more than 7 times than the other organs glucose oxygen need.
In brain metabolism, there is a high requirement for glucose / oxygen at steady rate.
The main ENERGY source is the generation of ATP by the aerobic metabolism Introduction Metabolism and CNS functions.
Many co-factors are required for the synthesis of the various neuro-transmitters, deficiencies of pyridoxal phosph, thiamine pyroph, and vitamin B12 result in a variety of neurologic dysfunctions.
There is a relationship between the oxidation of glucose and the supply of precursors for the synthesis of neurotransmitters in neurons within CNS.
Hypoglycemia (or hypoxia) affect brain function because they influence: ATP production for CNS neurons, Supply of precursors for neurotransmitter bio synthesis.
First clinical signs in hypoglycemia initiated by hypothalamic sensory nuclei as sweating, palpitations, anxiety and hunger, late CNS disorders: confusion/ lethargy/ seizures/coma.
At blood glucose falls below 45 mg/dL: the brain attempts to use internal substrates such as glutamate and TCA cycle intermediates as fuels for ATP production. Quickly depleted because the pool size of these substrates is quite small, they are quickly depleted.
When blood glucose: from 45 to 36 mg/dL NO EEG changes are observed, decreased synthesis of neurotransmitters. Symptoms appear to arise from decreased synthesis of neurotransmitters in particular regions of the brain like hippocampal and the cortical structures.
If blood glucose levels continue to fall below 18 mg/dL If blood glucose levels continue to fall below 18 mg/dL isoelectric EEG becomes isoelectric Neuronal cell death glutamate excitotoxicity Neuronal cell death ensues that may be caused by glutamate excitotoxicity (as result of ATP depletion) glutamate is the main excitatory neurotransmitter. Neurons that respond to glutamate are referred to as glutaminergic neurons Sources of glutamate in nerve terminals: Synthesized from glucose through glucose metabolism in neurones (main source) From blood (few as no cross BBB) Mechanism of action of glutamate as a neurotransmitter: Synthesis from glucose metabolism concentration in vesicles (in presynapses)2. Release by exocytosis to synaptic cleft Uptake by postsynaptic, Binding to glutaminergic receptors in postsynapses Functional effect.
Postsynaptic glutaminergic neurons perform their roles through: Ionotropic receptors 1- Ionotropic receptors that bind glutamate released from presynaptic neurons kainate referred to as kainate, 2-amino-3-hydroxy-5-methyl-4-isoxalone propionic AMPA NMDA acid (AMPA) and N-methyl-D-aspartate (NMDA) receptors. Metabotropic glutamate receptors 2- Metabotropic glutamate receptors that are members of the G-protein coupled receptor (GPCR) family.
Ionotropic Glutamate Receptors Non- NMDA, AMPA, KAINATE receptors generally allow the passage of Na + and K + NMDA receptors allows the passage of both Na + and Ca ++ ions. (More permeable to the Ca ++), role of astrocytes in glutamate metabolism in CNS: taken up astrocytes Glutamate released during neurotransmission is taken up primarily by neighboring astrocytes. Part of astrocyte glutamate is converted to glutamine by glutamine synthetase, which is abundant in astrocytes and absent from neurons. Glutamine is released from astrocytes and is taken up by neurons. In neurones, glutamine is deaminated to glutamate by glutaminase in neurons.
Glutamate Excitotoxicity is a pathological process by which nerve cells are damaged and killed by glutamate (prolonged opening of the receptor ion channel and the influx of lethal amounts of Ca2 ions, which can activate cytotoxic intracellular pathways in the postsynaptic neurons Glutamate Excitotoxicity Cerebral ischemia It is the potentially reversible altered state of brain physiology and biochemistry that occurs when substrate delivery is cut off or substantially reduced by vascular stenosis or occlusion.
Pathophysiology of Cerebral Ischemia ATP ↓↓ ATP Induction Amplification Expression.
Induction Phase Phase Lack of oxygen supply to ischemic neurones lactic acid the cell switches to anaerobic metabolism, producing lactic acid. ATP depletion malfunctioning of membrane ion system Depolarisation of neurones Influx of calcium glutamate Release of neurotransmitters as glutamate (glutamate excitotoxicity) (causing glutamate excitotoxicity) Accumulation of more intracellular levels of Ca2+ which causes additional release of glutamate (viscious cycle) Amplification Phase Expression Phase 1-overexcites cells and causes the generation of harmful chemicals like free radicals (causing oxidative stress) free radicals (causing oxidative stress) 2- Activation of calcium-dependent enzymes such as: calpain (causing apoptosis) phospholipases (causing membrane breakdown) phospholipases (causing membrane breakdown) glutamate excitotoxicity 3- Calcium can also cause the release of more glutamate (glutamate excitotoxicity) phospholipases The cell’s membrane is broken down by phospholipases Cell membrane becomes more permeable, and more ions and harmful chemicals flow into the cell. + apoptotic Mitochondria membrane break down, releasing toxins and apoptotic factors into the cell Ca 2+ lactic acid is produced in excess in ischemia In cerebral ischemia, lack of oxygen switches metabolism of glucose to the anaerobic pathway & lactic acid production Lactic acid contribute to the pathophysiology of ischemia as Lactic acid contribute to the pathophysiology of ischemia as: It decreases pH that may injure and inactivate mitochondria 1- It decreases pH that may injure and inactivate mitochondria. Lactic acid degradation of NADH 2- Lactic acid degradation of NADH (which is needed for ATP synthesis) may also interfere with adequate post-ischemic recovery of ATP levels. Lactic acid increase the amount of free-radical mediated injury 3- Lactic acid increase the amount of free-radical mediated injury. Lactic acid in neurons acidosis promotes the pro-oxidant effect ↑ the rate of conversion of O 2 - to H 2 O 2 or to hydroxyl-peroxyl radical Reactive oxygen species are formed from partial reduction of molecular O 2 i.e. adding electrons to oxygen leading to the formation of superoxide, hydrogen peroxide and hydroxyl radical. ROS cause damage to DNA, protein and unsaturated lipids of the cells Generally, ROS cause damage to DNA, protein and unsaturated lipids of the cells. What is meant by oxidative stress ROS antioxidants a condition in which cells are subjected to excessive levels of ROS and they are unable to counterbalance their deleterious effects with antioxidants Oxidative stress is caused by ischemia?
Oxidative stress
Why the Brain is vulnerable to Oxidative Stress. High metabolic requirement for oxygen; Prevalent concentration of oxidizable polyunsaturated fatty acids (PUFAs) in membranes; Enrichment in redox active metals, most notably iron The presence of potentially phagocytic microglia which can be a major source of ROS and free radicals when activated by injury.
Oxidative stress Cellular Effects of Reactive Oxygen Species in CNS Nitric oxide is over produced and turns to be a neurotoxic mediator as it reacts with superoxide anions to generate toxic peroxynitrite which leads to production of more potent neurotoxin such as hydroxyl radicals Lipid peroxidation Lipid peroxidation Inactivation of enzymes Inactivation of enzymes Nucleic acid (DNA, RNA) damage Nucleic acid damage Release of calcium ions from intracellular stores with more damage to neurones Release of calcium ions from intracellular stores with more damage to neurons.
Damage to cytoskeleton
Apoptosis and necrosis are caused by ischemia
Necrosis: is commonly observed early after severe ischemic insults Apoptosis Apoptosis: occurs with more mild insults and with longer survival periods calpain- mediated proteolysis The mechanism of cell death involves calcium-induced calpain- mediated proteolysis of brain tissue Substrates for calpain include: – Cytoskeletal proteins – Membrane proteins – Regulatory and signaling proteins.
Stroke Mitochondria break down, releasing toxins and apoptotic factors into the cell. The caspase-dependent apoptosis cascade is initiated, causing cells to “commit suicide.”
Stroke Caplains are cytosolic proteinases whose irreversible proteolytic activity is against cytoskeleton and regulatory proteins and similar substances). Over activated (overstimulated).
When receptors for glutamate such as the NMDA and AMPA receptor are over activated Excessive excitation of glutamate receptors has been associated with hypoglycemia and stroke (cases in which there is lack of glucose or oxygen ending in lack of energy production in CNS).
According Tapia R., “ALS was described first in 1869 by Charcot, is a devastating neuro-degenerative disease characterized by the selective and progressive loss of upper and lower motor neurons of the cerebral cortex, brainstem and the spinal cord. The result of this loss is a progressive and irreversible paralysis leading to a complete incapacity of movements and finally to respiratory failure, but the cognitive functions are not affected and is not merely the result of aging because may occur at young adult ages. There is still no treatment, prevention or reliable biomarkers of ALS [59].
Although progress in neurochemical, physiological, genetic and clinical investigations in the last decades has identified several cellular processes and mechanisms that seem to be involved in the neuronal death, such as glutamate receptors-mediated excitotoxicity, disruption of spinal inhibitory circuits, inflammatory events, axonal transport deficits, oxidative stress, mitochondrial dysfunction, energy failure, intracellular Ca2+ dishomeostasis, protein aggregation and misfolding, changes in gene expression, astrocytes alterations, and non-cell autonomous toxic factors, the understanding of the origin and temporal progress of the disease is still incomplete and insufficient. There is a need of further experimental models and approaches to discern the importance of such mechanisms and to discover the factors that determine the selective death of motor neurons characteristic of ALS, in contrast to other neurodegenerative diseases such as Parkinson’s and Alzheimer’s disease in which other neuronal types located in other CNS regions are predominantly affected.
The only known cause of the ALS disease is associated with genetic mutations, mainly in the gene encoding superoxide dismutase 1 (SOD1), the most frequent case of familial ALS (FALS there is no known cause of the sporadic form of ALS (SALS), that comprises >90% of all cases, although several other mutations have recently been identified also in SALS. Both ALS types show similar histopathological and clinical characteristics, and in spite of numerous investigations using tissue from ALS patients and relevant advances in the design of several experimental models of motor neuron degeneration, it has not been possible to establish a clear cause-effect relationship regarding the loss of motor neurons or the motor alterations characteristic of the disease, as fasciculations, spasticity, and progressive paralysis.
Studies in vitro in cell cultures, tissue slices or organotypic preparations have given useful information regarding the cellular and molecular mechanisms of motor neuron death. except for transgenic rodents expressing mutant forms of human SOD1 (mSOD1), which are the most extensively used model in vivo in spite of the low frequency of FALS as compared to SALS, experiments in living animals are scarce.
The role of astrocytes and microglia from mSOD mice in non-cell autonomous processes, including the release of toxic factors, is studied by Rojas et al. and Trias et al., with a useful ex vivo approach.
Staats et al., Bowerman et al., show the involvement of microglia and neuroimmunological events in the progress of ALS and suggest possible therapeutic strategies.
The molecular modifications of mSOD1 protein, and how such alterations, like oxidation, aggregation or misfolding, result in its toxicity, are reviewed by Furukawa; Rotunno and Bosco, and Ogawa and Furukawa. Again in mSOD1 mice but with a different approach, Mòdol et al., show a possible role of the potassium chloride cotransporter 2 in spasticity, including results on the role of spinal inhibitory interneurons.
A partial protective effect of the NMDA receptor antagonist gacyclidine on the motor deficits in mSOD1 mice is shown by Gerber et al., and the role of trophic factors in the modulation of motor neurons activity as well as their protective effect in several models of ALS, is reviewed by Tovar-y-Romo et al., and Lladó et al. It is emphasized in particular the potent protective action of VEGF.
A general panorama of the role of lipids in ALS is presented by Schmitt et al., with emphasis on energy and signaling mechanisms. Garbuzova-Davis and Sanberg review the vascular changes that may alter blood brain barrier and thus be involved in the pathogenesis of ALS, as compared to the transgenic rodent models.
Why the clinical manifestations of motor neuron death in both FALS and SALS appear in young adulthood but are not age-related, particularly the former, in spite of the fact that genetic alterations are present since the beginning of individual life?
Why the motor neuron degeneration progresses rapidly after the initial symptoms appear?
It is now clear that the loss of motor neurons in ALS is multifactorial, but many of the deleterious factors seem to occur also in other neurodegenerative diseases like Parkinson’s and Alzheimer’ disease; then, maybe the crucial question to be answered is: what determines the predominant, if not totally selective characteristic death of motor neurons in ALS, sparing other neuronal types?
Because we have no indication or biomarker of the susceptibility to ALS and of when will it manifest, it is extremely difficult to prevent or treat the disease.
When symptoms have already started and the diagnosis is made it is probably too late to impede the progression of neuronal degeneration. A verifiable resolution of the above questions seems a requisite for efficient and reliable treatments or prevention of this dreadful disease.
Is possible to see various form: sporadic, familiar, west pacific form. Juvenile form with intracityoplasmatic inclusion and other.
Starting from the characteristic geographic presentation of ALS is interesting to verify some Relationship to better clear the pathological movens in this kind of complex disorder.
Many studies show a specific neurotoxicity of Annonaceous acetogenins from edible Annona fruits.
Also is cleared that rotenone toxicology results in α-synuclein aggregates in the presence of rotenone and non-aggregated α-synuclein in midbrain axons of MPTP-treated squirrel monkeys.
Even if is not clearly associated acetogenin from diet as neurotoxic factor as some ALS /PD form direct causal agent an artificial subministration of this kind of subtances result in final motor neuron inclusion and aggregates (as see α-synuclein aggregates in rotenone).
This fact can be considered a toxicological model to show aggregates productions after a toxic event in some neurons.
(The increasing accumulation of bioactive derivates of agetogenin, often deleterious molecules through higher trophic levels of a food chain can better explain this toxicological process.)
Exposure to the neurotoxic non-protein amino acid β-N-methylamino-L-alanine (BMAA) may play a role in neurodegenerative processes.
BMAA is produced across the cyanobacterial order and it has been detected in several aquatic and terrestrial environments worldwide, suggesting that it is ubiquitous.
Other examples useful in this work on neuronal toxicity can be neurolatirism disease and the rotenone toxicity.
Strong evidence indicates that pesticides play a role in ALS.
Studies show also Occupational exposure to ELF-EMF was significantly associated with increased risk of ALS in pooled studies (RR = 1.29, 95%CI = 1.02-1.62), and case-control studies (OR = 1.39, 95%CI = 1.05-1.84), but not cohort studies (RR = 1.16, 95% CI = 0.80-1.69).
ALS of ischemic origin in the intraparenchymal territory of the AVSAs and/or in anterior spinal artery may be related by atherosclerosis and associated to anatomical variants in the V4 segments of the vertebral arteries.
All this example show a pathological moves that results in a final damages.
ALL this evidence in this work are then compared with the unic geographic cluster presentation of this pathology (Figures 7-12).
Figure 7: Some neurone differecnces (From literature).
Figure 8: EEG of the patient: Periodic discharges and focal slowing in the left fronto-temporal areas, visible in an early stage of the HSV-1 encephalitis.
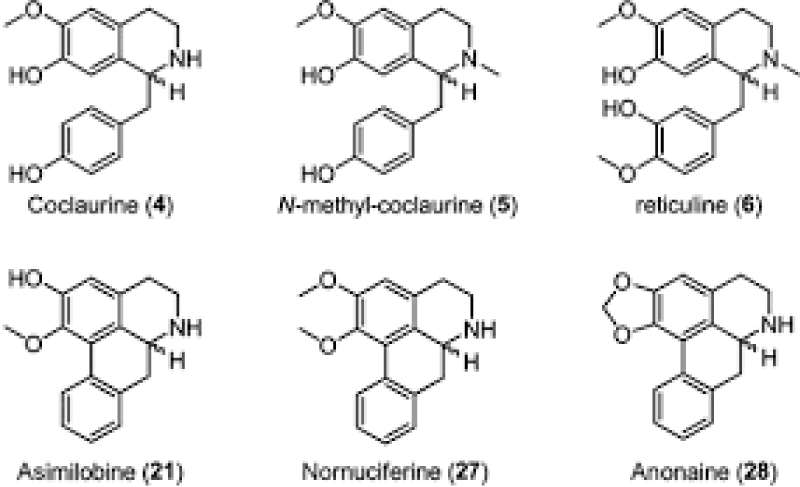
Figure 9: ANNONAINE structure.
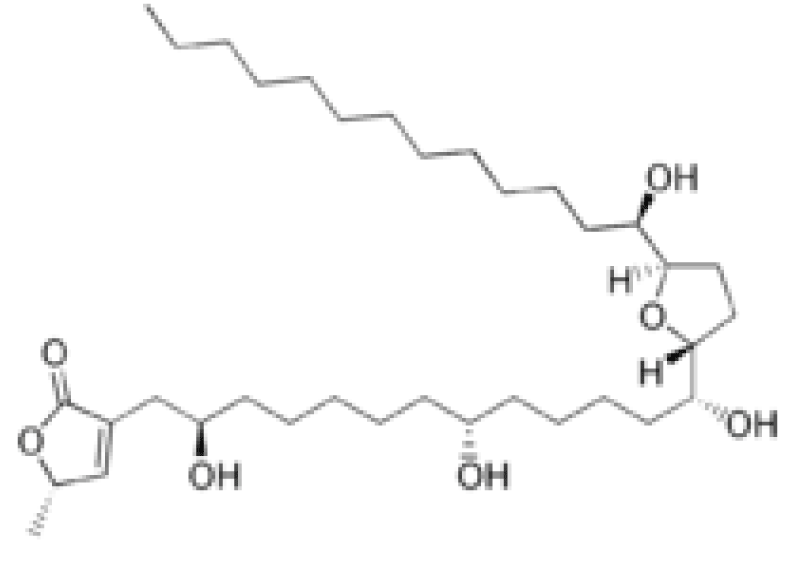
Figure 10: ANNONACIN structure.
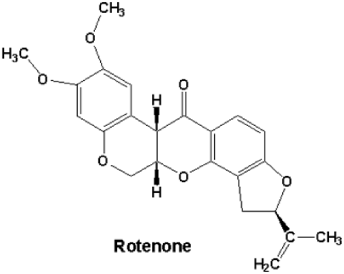
Figure 11: Rotenone.
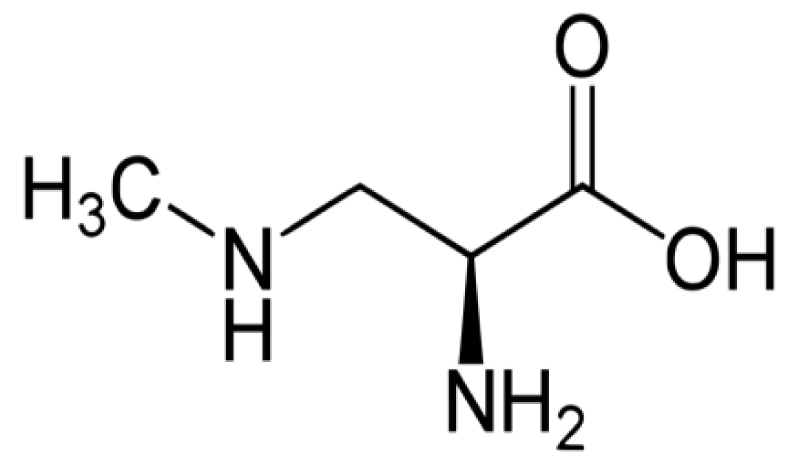
Figure 12: β-Methylamino-L-alanine, or BMAA, is a non-proteinogenic amino acid produced by cyanobacteria.
“A neurodegenerative tauopathy endemic to the island of Guadeloupe has been associated with the consumption of anonaceous plants that contain acetogenins, potent lipophilic inhibitors of complex I of the mitochondrial respiratory chain. To test the hypothesis that annonacin, a prototypical acetogenin, contributes to the etiology of the disease, we investigated whether annonacin affects the cellular distribution of the protein tau. In primary cultures of rat striatal neurons treated for 48 h with annonacin, there was a concentration-dependent decrease in ATP levels, a redistribution of tau from the axons to the cell body, and cell death. Annonacin induced the retrograde transport of mitochondria, some of which had tau attached to their outer membrane. Taxol, a drug that displaces tau from microtubules, prevented the somatic redistribution of both mitochondria and tau but not cell death. Antioxidants did not affect either the redistribution of tau or cell death. Both were prevented, by forced expression of the NDI1 nicotinamide adenine dinucleotide (NADH)-quinone-oxidoreductase of Saccharomyces cerevisiae, which can restore NADH oxidation in complex I-deficient mammalian cells and stimulation of energy production via anaerobic glycolysis. Other ATP-depleting neurotoxins (1-methyl-4-phenylpyridinium, 3-nitropropionic, and carbonyl cyanide m-chlorophenylhydrazone) reproduced the somatic redistribution of tau, whereas toxins that did not decrease ATP levels did not cause the redistribution of tau. Therefore, the annonacin-induced ATP depletion causes the retrograde transport of mitochondria to the cell soma and induces changes in the intracellular distribution of tau in a way that shares characteristics with some neurodegenerative diseases” [1] (Figure 13).

Figure 13: Annona muricata L.
N Bonneau et al., “Annonaceous acetogenins are inhibitors of the first complex in mitochondrial respiratory chain. They are proposed as environmental neurotoxins responsible for Guadeloupean atypical Parkinsonism. The edible fruits of Annona muricata L. (soursop) and Asimina triloba Dunal (pawpaw) were previously shown to constitute major sources of exposure to these compounds. We performed study of total extracts using MALDI-TOF MS and dereplicative methods, and here propose quantitative data showing homogeneity between fruits of A. muricata from diverse origins, in which annonacin is the major AAG. The fruits of A. squamosa also constitute a source of exposure, with squamocin as the major representative of AAGs. Our results are significant in regard to public health and support the inclusion of Annona spp. exposure/consumption in studies investigating potential risk factors for neuro-degeneration”[2].
Lannuzel A et al., “In the French West Indies there is an abnormally high frequency of levodopa-resistant parkinsonism, suggested to be caused by consumption of fruit and infusions of tropical plants, especially Annona muricata (corossol, soursop). To determine whether toxic substances from this plant can cause the neuronal degeneration or dysfunction underlying the syndrome, we exposed mesencephalic dopaminergic neurons in culture to the total extract (totum) of alkaloids from Annona muricata root bark and to two of the most abundant subfractions, coreximine and reticuline. After 24 hours, 50% of dopaminergic neurons degenerated with 18 microg/ml totum, 4.3 microg/ml (13 microM) coreximine, or 100 microg/ml (304 microM) reticuline. The effects of the alkaloid totum were not restricted to the population of dopaminergic cells since GABAergic neurons were also affected by the treatment. Nuclei in dying neurons showed DNA condensation or fragmentation, suggesting that neuronal death occurred by apoptosis. Cell death was not excitotoxic and did not require toxin uptake by the dopamine transporter. Neuro-degeneration was attenuated by increasing the concentration of glucose in the culture medium, which also reduced the effect of the dopaminergic neurotoxin MPP+, a mitochondrial respiratory chain inhibitor. Toxin withdrawal after short-term exposure arrested cell death. Acute treatment with totum, coreximine, or reticuline reversibly inhibited dopamine uptake by a mechanism that was distinct from that causing neuronal death. GABA uptake was not reduced under the same conditions. This study suggests that alkaloids from A. muricata can modulate the function and the survival of dopaminergic nerve cells in vitro. It is therefore conceivable that repeated consumption could cause the neuronal dysfunction and degeneration underlying the West Indian parkinsonian syndrome” [3].
Höllerhage M et al., “Dietary supplements containing plant materials of Annonaceae species. Were extracted by hot, pressurized ethyl acetate and analyzed for their effect in vitro on Lund human mesencephalic neurons. Cell viability was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, and cell death was determined by lactate dehydrogenase levels. Three supplements strongly decreased the cell viability at extract concentrations of 1 µg/mL, of which 1 decreased cell viability at 0.1 µg/µL. Also, strong neuronal toxicities of these supplements were found. Cell death was observed at concentrations of 10 µg/mL. The degree of toxicity was comparable to the ones found in Annonaceous fruit extracts. 2 fruit pulps of Annonaceae (A. muricata and A. squamosa) showed a reduction in cell viability at lower concentrations. The fruit pulp extract of A. muricata revealed the strongest neurotoxic effect, with 67% cell death at a concentration of 1 µg/mL. A high reduction in cell viability coupled with pronounced cell death was found at 0.1 µg/mL for an Annonaceous seed extract. These results demonstrate that the intake of dietary supplements containing plant material from Annonaceae may be hazardous to health in terms of neurotoxicity” [4].
Zafra-Polo MC et al., “One-hundred and twenty-eight different linear, epoxy, mono-tetrahydrofuran, bis-tetrahydrofuran or tri-tetrahydrofuran acetogenins have been isolated from the Annonaceae. These new secondary metabolites are potent cytotoxic inhibitors of the mitochondrial NADH: ubiquinone oxidoreductase (complex I of the respiratory chain)” [5].
Effects of ethanol extract of Annona muricata on pentylenetetrazol‐induced convulsive seizures in mice.
P. N’Gouemo et al., “The effects of an ethanol extract of the mature leaves of Annona muricata (Annonaceae), were investigated on pentylenetetrazol (PTZ)‐induced clonic‐tonic seizures in mice. The latency of clonic PTZ seizures and the incidence of clonic and tonic episodes of PTZ seizures were analyzed. The results demonstrated that the plant extract significantly reduced the incidence of tonic PTZ seizures and mortality, and lengthened the onset to clonic PTZ seizures. This indicates that extract of the leaves of A. muricata contains active substance(s) that preferentially suppressed the tonic phase of PTZ‐induced clonic‐tonic seizures” [6].
Moghadamtousi SZ et al., “Annona muricata, is commonly known as soursop and graviola. The leaves of this tropical fruit tree are widely used in folk medicine against skin diseases and abscesses, there is no scientific evidence justifying the use of A. muricata leaves. The aim of this study is to evaluate the wound healing potential of ethyl acetate extract of A. muricata leaves (EEAM) towards excisional wound models in rats.
Sprague Dawley rats (24) were randomly divided into four groups, viz. (A) vehicle control, (B) low dose of EEAM (5% w/w), (C) high dose of EEAM (10% w/w) and (D) positive control with excisional wound created on the neck area. Wounds were topically dressed twice a day for 15 days. On the 15th day, animals were sacrificed and then processed for immune-histochemical and histological evaluations, including Hematoxylin - Eosin and Masson Trichrome stainings. The activity of antioxidants, namely catalase, glutathione peroxidase and superoxide dismutase, and malondialdehyde (MDA) was measured in wound tissue homogenate.
Macroscopic - microscopic analysis of wounds demonstrated a significant wound healing activity shown by EEAM at 2 doses. Treatment of wounds with ointment containing EEAM caused significant surge in antioxidants activities and decrease in the MDA level of wound tissues compared with vehicle control. The immunohistochemical evaluation revealed conspicuous up-regulation of Hsp70 in treated wounds with EEAM, suggesting the anti-inflammatory effect of EEAM.
EEAM exhibited a promising wound healing potential towards excisional wound models in rats” [7].
Chen Y et al., “Annonaceous acetogenins are one of the most interesting classes of natural products appearing in the past two decades. Here, we studied the antitumor activity and toxicity relationship of ACGs including annosquamin B (1), bullatacin (2) and annosquatin B (3) in vivo. A single intraperitoneal (i.p.) injection of 100 μg/kg of annosquamin B, bullatacin and annosquatin B did not cause side effects in normal mice. Bullatacin treatment with five doses of 25 and 50 μg/kg in H22 hepatoma cells bearing mice resulted in about 61% reduction in tumor growth with hematologic parameters increased significantly in normal mice. Annosquamin B and annosquatin B treatments with 10 doses of 25, 50 and 100 μg/kg in the H22 hepatoma cells transplantation tumor model mice resulted in maximum 53.7% and 58.7% reduction in tumor growth, respectively, and did not cause severe side effects in normal mice. This study provided the evidence that adjacent bis-THF ACGs showed higher antitumor activity and toxicity than mono-THF and nonadjacent bis-THF ACGs in vivo. it was found that bullatacin led to liver and kidney toxicity via increasing calcium concentration, ROS production, and Bax expression and Bax/Bcl-2 ratio in rats with repeated treatment with bullatacin for 3 weeks” [8].
Ribeiro Ldo P et al., “Acetogenins, a class of natural compounds produced by some Annonaceae species, are potent inhibitors of mitochondrial electron transport systems. Although the cellular respiration processes are an important biochemical site for the acaricidal action of compounds, few studies have been performed to assess the bioactivity of acetogenin-based biopesticides on spider mites, mainly against species that occur in orchards. ESAM is a homemade biopesticide which was previously characterized by its high concentration of acetogenins. It caused both high mortality of P. citri females (LC50 = 7,295, 4,662, 3,463, and 2,608 mg l (-1), after 48, 72, 96, and 120 h of exposure, respectively) and significant oviposition deterrence (EC50 = 3.194,80 mg l(-1)). There was no effect on P. citri female fertility (hatching rate). In addition, the ESAM efficacy (in terms of its LC90) was compared with commercial acaricides/insecticides (at its recommended rate) of both natural [Anosom(®) 1 EC (annonin), Derisom(®) 2 EC (karanjin), and Azamax(®) 1.2 EC (azadirachtin + 3-tigloylazadirachtol)] and synthetic origin [Envidor(®) 24 SC (spirodiclofen)]. Based on all of the analyzed variables, the ESAM exhibited levels of activity superior to other botanical commercial acaricides and similar to spirodiclofen. Our results indicate that ESAM may constitute a biorational acaricide for citrus red mite integrated pest management in Brazilian citrus orchards, particularly for local use” [9].
Levine RA et al., “The concentrations of the neurotoxins, annonacin and squamocin, were determined in a lyophilized sample of the fruit pulp of the North American pawpaw (Asimina triloba) by LC coupled to high resolution mass spectrometry or LC-HRMS. Extracted using dry methanol at 100 °C and 10 MPa pressure in a sealed container. The extraction of annonacin and squamocin was optimal at 100 °C with 7.72 and 0.162 mg/g, respectively, being found. Several isomers of annonacin and squamocin were separated and detected but not quantified” [10].
Zhen Jiang et al., “ALS is the most common motor neuron disease characterized by progressive loss of motor neurons in the brainstem and spinal cord. There is no cure or effective treatment for ALS and the cause of disease is unknown in the majority of ALS cases. Neuronal mitochondria dysfunction is one of the earliest features of ALS. Mitochondria are highly dynamic organelles that undergo continuous fission, fusion, trafficking and turnover, all of which contribute to the maintenance of mitochondrial function. Abnormal mitochondrial dynamics have been repeatedly reported in ALS and increasing evidence suggests altered mitochondrial dynamics as possible pathomechanisms underlying mitochondrial dysfunction in ALS” [11].
Ravindranath V, “beta-N-Oxalyl amino-L-alanine (L-BOAA); synonym beta-N-oxalyl-alpha, beta-diaminopropionic acid (beta-ODAP) is a naturally occurring non-protein amino acid present in the chickling pea from the plant Lathyrus sativus grown in drought prone areas. Ingestion of L-BOAA as a staple diet results in a progressive neurodegenerative condition, neurolathyrism, a form of motor neuron disease which affects the upper motor neurons and anterior horn cells of the lumbar spinal cord. L-BOAA is an excitatory acid and acts as an agonist at the AMPA receptor. One of the primary effects of L-BOAA toxicity is the inhibition of mitochondrial complex I selectively in the motor cortex and lumbar spinal cord. Evidence has suggested that the mitochondrial dysfunction is a consequence of oxidation protein thiol groups as a result of generation of reactive oxygen species. Mitochondrial complex I is highly too vulnerable to inactivation through oxidation of vital sulfhydryl groups. Thiol antioxidants such as alpha-liopic acid offer a method of protecting mitochondrial function. A common mechanism involving oxidation of protein thiol groups may underlie neurodegeneration occurring through mitochondrial dysfunction induced by excitatory amino acid” [12].
Lisa F et al., “The acetogenin, annonacin, from the tropical annonaceous plant Annona muricata, is a lipophilic, mitochondrial complex I inhibitor reported to be more toxic than rotenone to mesencephalic neurons. The temperate annonaceous plant Asimina triloba (pawpaw) is native to the Eastern United States and products are available online.
Pawpaw extract was prepared by pulp extraction with methanol and liquid–liquid partitioning with ethyl acetate (EtOAc). Annonacin was isolated from the crude EtOAc extract via column chromatography using a gradient solvent system of increasing polarity. Mass spectroscopy, nuclear magnetic resonance and infrared spectroscopy were used to compare isolated material with synthetic annonacin data and a natural annonacin sample. Toxicity of isolated annonacin and the total EtOAc extract was determined in primary rat cortical neurons using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay.
The average concentration of annonacin in the fruit pulp was 0.0701 ± 0.0305 mg/g. Purified annonacin (30.07 μg/ml) and crude EtOAc extract (47.96 μg/ml) induced 50% death of cortical neurons 48 h post treatment. Annonacin toxicity was enhanced in the presence of crude extract.
Pawpaw fruit contains a high concentration of annonacin, which is toxic to cortical neurons. Crude fruit extract also induced neurotoxicity, highlighting the need for additional studies to determine the potential risks of neurodegeneration associated with chronic exposure to pawpaw products” [13].
Panov A et al., “Chronic infusion of rotenone (Rot) to Lewis rats reproduces many features of Parkinson disease. Rot (3 mg/kg/day) was infused subcutaneously to male Lewis rats for 6 days using Alzet minipumps. Control rats received the vehicle only. Presence of 0.1% bovine serum albumin during the isolation procedure completely removed rotenone bound to the mitochondria. Therefore all functional changes observed were aftereffects of rotenone toxicity in vivo. In Rot rat brain mitochondria (Rot-RBM) there was a 30-40% inhibition of respiration in State 3 and State 3U with Complex I (Co-I) substrates and succinate. Rot did not affect the State 4Deltapsi of RBM and rat liver mitochondria (RLM). Rot-RBM required two times less Ca2+ to initiate permeability transition (mPT). There was a 2-fold increase in O*2- or H2O2 generation in Rot-RBM oxidizing glutamate. Rot infusion affected RLM little. Our results show that in RBM, the major site of reactive oxygen species generation with glutamate or succinate is Co-I. We also found that Co-II generates substantial amounts of reactive oxygen species that increased 2-fold in the Rot-RBM. Our data suggest that the primary mechanism of the Rot toxic effect on RBM consists in a significant increase of O*2- generation that causes damage to Co-I and Co-II, presumably at the level of 4Fe-4S clusters. Decreased respiratory activity diminishes resistance of RBM to Ca2+ and thus increases probability of mPT and apoptotic cell death. We suggest that the damage to Co-I and Co-II shifts O*2- generation from the CoQ10 sites to more proximal sites, such as flavines, and makes it independent of the RBM functional state” [14].
Wei-Bin Shen et al., “The tragic development of a significant cluster of cases of ALS/PDC among the Chamorro population of Guam generated much interest as to a common etiology for these neurodegenerative disorders. The ingestion of washed flour prepared from seeds of the plant Cycas micronesica during a period of famine has been implicated. This is not without precedent. In Guadeloupe atypical Parkinsonism has been linked to the consumption of the fruit and leaves of the tropical plant Annona muricata (soursop).Patients present with symptoms of progressive supranuclear palsy or Guadeloupian PDC with cerebral atrophy. Pathologically, there is neurodegeneration of both cholinergic cells and GABAergic cells of the STR and cholinergic and DAergic cells of the substantia nigra, differing from our observations with cycad exposure both in vivo - in vitro.
We began our investigation of cycad-induced neuro-degeneration in an attempt to develop another rat model of ALS. In cycad-fed mice, Wilson and Shaw demonstrated an ALS-type syndrome with later denervation of DAergic neurites in the STR. we found parkinsonian behavioral symptoms in cycad-fed rats including slowness of movement, tremulous jaw movements, a shuffling gait, unilateral rotations and increased grip strength. We conducted neuro-chemical and histological analysis of the brains of cycad-fed rats and found that cycad ingestion causes a progressive loss of DAergic innervation of the STR, and progressive degeneration of nigral DAergic neurons later with the remaining DAergic neurons showing ubiquitin aggregates and proteinase K-resistant aggregates of phospho α-syn. We also found phospho α-syn aggregates in the locus coeruleus and cingulate cortex of cycad-fed rats (Supplementary, S4) as has been reported in PD. Cycad ingestion causes a slow progressive loss of DAergic neurons rather than the rapid loss seen in other toxin-induced models of PD; and cycad ingestion also causes α-synuclein aggregates colocalized with ubiquitin, a common marker in PD. In the Dopamine receptor 2 (D(2)R) knockout mouse, cytoplasmic inclusions containing α-synuclein and ubiquitin were present in the SNc neurons of older D(2)R(−/−) mice, and were also occasionally seen in aged wild-type mice. The phosphorylated α-synuclein antibody used in our studies binds to α-synuclein, phosphorylated at residue Ser-129, a post-translational modification shown to be a toxic form of the protein and indicative of increasing levels of oxidative stress in these cells. In cycad-fed rats, there was a time-dependent accumulation of aggregates of α-synuclein first in DAergic neurites in the STR and later in DAergic neurons in the SNc. This loss of the TH-ir neurons in the SNc could be a loss of the TH phenotype in the absence of an actual cell loss of DAergic neurons. Our stereological counts of TH-ir neurons at 51 weeks of age (Cohort 1 B) showed a significant loss of TH-ir neurons in the presence of increased α-synuclein aggregates. Chu and Kordower found in PD patients and in non-human primates that increases in α-synuclein are age-associated and we found a similar phenomenon in older cycad-fed rats. Rats in our cohorts did not develop a loss of motor neurons in the lumbar cord or increased gliosis in the grey matter of the spinal cord.
Organotypic nigrostriatal co-cultures offer a useful tool for studying dopaminergic degeneration in a natural circuit. These slice cultures survive over two months, making chronic intoxication studies feasible. Chronic treatment with an organic extract of washed cycad over two weeks resulted in an 85% decrease of TH-ir somata in the SN, either by loss of TH-ir neurons or a loss of the TH phenotype. The lack of NeuN-ir cell loss in the SN from slice cultures is possibly due to loss of the TH phenotype in the absence of actual TH-ir cell loss. The abnormal morphology of the TH-ir neurons in the presence of cycad suggests that death of DAergic neurons, which are a small number of the total number of NeuN-ir neurons, may occur. In the STR of slice cultures, there is a small (not- significant) loss of NeuN-ir cells which is clearly distinct from the 85% loss of TH-ir cells in the SN. Possible explanation is that the concentrations of cycad toxins in the slice culture differ from the dose of cycad ingested in vivo, leading to slightly different selectivity. phospho α-syn accumulation in TH-ir neurons and neurites in the NSOC was observed. This preparation is the first demonstration of phospho α-syn aggregates coupled with a loss of DAergic neurons in an organotypic slice without a genetic alteration.
A number of natural and synthetic molecules exert deleterious effects on DAergic neurons including: 6-hydroxydopamine (6-OHDA), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), rotenone, and paraquat.
None of these toxins causes a progressive syndrome that closely resembles idiopathic Parkinsonism in humans. Brains of individuals who died after MPTP exposure showed evidence of DA neuronal loss in the SNc in the absence of Lewy bodies in the remaining neurons.
The temporal loss of DAergic innervation in the STR before the loss of DAergic neurons in the SNc in cycad-fed rats mimics a similar phenomenon in non-human primates in a chronic low dose MPTP model of PD. Braak et al., have proposed that sporadic PD can be staged in the brain based upon nonrandom neuronal dysfunction and Lewy body pathology.
To demonstrate similarities between cycad model and sporadic PD we investigated whether aggregates of α-synuclein occur in other regions of the brain in addition to the SNc.
We found these aggregates in the locus coeruleus and cingulate cortex, similar to sporadic PD. Previous studies have found both α-synuclein aggregates in the presence of rotenone and non-aggregated α-synuclein in midbrain axons of MPTP-treated squirrel monkeys. Our in vivo findings in the cycad-fed rat makes this model an interesting addition to the study of the relationship between α-synuclein aggregates and DAergic neurodegeneration in a non-genetic model, recapitulating some of the progressive behavioral and histopathological abnormalities seen in the human disorder. Unlike PDC, the cycad-fed rat showed no pathological signs of NFTs in the cortex. Thus the rat exposed to cycad flour has both similarities (Parkinsonism) and differences (synuclein aggregates in the SNc and no cortical NFTs) with pathology found in patients with PDC.
The mechanism of neurotoxicity behind cycad consumption is unknown. One environmental interaction that can affect neurons is exposure to high levels of phytosterols and sterol glucosides. Plant sterols enter the mammalian body only via the diet. Relatively large amounts of plant sterols are present in plant oils, nuts, and avocados, and interestingly, cycad flour. Phytosterols have a comparable chemical structure to cholesterol and can accumulate in the brain.In addition, phytosterols such as sitosterol and campesterol can activate Liver X Receptors (LXRs). Activation of LXRs have an important role in cholesterol metabolism and it has been determined that cholesterol metabolites, like 24-hydroxycholesterol, and certain phytosterols, are ligands for the LXR-β receptor.
Administration of β-sitosterol to LXR-β −/− mice accelerated the previously observed motor neuron degeneration in this knockout, and causes a loss of DAergic neurons in the SNc. β-sitosterol also reduced brain cholesterol levels in not only the LXR-β −/− mouse but in wild type mice as well. These studies suggest that LXR-β signaling is abnormal in the presence of β-sitosterol, leading to neuronal damage in the SNc.
Thus phytosterols and sterol glucosides could be etiological agents in PD and our current studies are focusing on this intriguing avenue in an attempt to elucidate the mechanism of cycad toxicity.
Unlike genetic models or toxicant based end-stage models of Parkinsonism that may have a great deal of phenotypic similarity among animals, there is considerable variability in the response of individual animals to cycad ingestion. This variability may be due to first-pass metabolism of cycad flour or due to the unevenness of uptake of phytosterols and other components of cycad flour into the brain.
We believe that the novelty of this cycad-fed rat model is that it recapitulates the variability and progression of symptoms and neuropathology seen in human Parkinsonism. It also allows examination of the early stages of the disorder. We believe that the cycad model in rats can uniquely serve as a progressive model of Parkinsonism” [15].
Laura Louise Scott et al., “Cyanobacterial β-N-methylamino-l-alanine (BMAA) has been suggested as a causative or contributory factor in the development of several neurodegenerative diseases. No BMAA animal model has adequately shown clinical or behavioral symptoms that correspond to those seen in either AD, ALS or PD. We present here the first data that show that when neonatal rats were exposed to BMAA on postnatal days 3, 4 and 5, but not on gestational day 14 or postnatally on days 7 or 10, several AD and/or PD-related behavioral, locomotor and cognitive deficits developed. Male rats exhibited severe unilateral hindlimb splay while whole body tremors could be observed in exposed female rats. BMAA-exposed rats failed to identify and discriminate a learned odor, an early non-motor symptom of PD, and exhibited decreased locomotor activity, decreased exploration and increased anxiety in the open field test. Alterations were also observed in the rats’ natural passive defense mechanism, and potential memory deficits and changes to the rat’s natural height avoidance behavior could be observed as early as PND 30. Spatial learning, short-term working, reference and long-term memory were also impaired in 90-day-old rats that had been exposed to a single dose of BMAA on PND 3–7. These data suggest that BMAA is a developmental neurotoxin, with specific target areas in the brain and spinal cord. The cyanobacterial metabolite β-N-methylamino-l-alanine (BMAA), found in many commonly consumed food items, has been implicated in the development of the neurodegenerative diseases (ALS/PDC), (ALS) and (AD). This proposed link is based largely on the high concentration of the amino acid in the diet of the Chamorro people of Guam, who had an extremely high incidence of ALS/PDC, and on the detection of BMAA in the brain tissue of Chamorro ALS/PDC, Canadian AD, sporadic ALS and Parkinson’s Disease (PD) patients. to date no animal model of BMAA toxicity has successfully shown clinical or behavioral symptoms together with neuropathology, that correspond to symptoms observed in patients with these neurodegenerative diseases.

Figure 14: Cyanobacteria BMAA.
Although acute toxicity of BMAA, when administered at doses as high as 4000 mg per kilogram body weight has been shown in chicks, rats, and mice and some neuro-toxic effects such as hind leg splay and rigidity have been described when BMAA is administered via intra-cerebroventricular injections, studies using systemic administration of an environmentally relevant BMAA dose to adult animals have not been successful in linking BMAA to neuro-degenerative diseases. Polsky et al., Perry et al., Duncan et al., Cruz-Aguado et al., and Scott et al. did not observe any clinical signs of toxicity associated with the chronic administration of BMAA to adult rats, and only Seawright et al., were able to show some neurodegeneration with chronic high dose exposure (1000 mg BMAA/kg body weight per day). Karlsson et al., attributed the lack of symptoms observed in adult rats to the low transport of BMAA into the adult rodent brain and subsequently showed a substantially higher transfer of BMAA into the brain of neonatal rats with distinct localization of the neurotoxin in the hippocampus and striatum. Additionally, Smith et al., demonstrated that BMAA gains access to the brain via the cerebrovascular large neutral amino acid transporter and consequently suggested that modulators of brain neutral amino acid uptake, such as the observed enhanced uptake rate in infants, would similarly modulate the transport of BMAA into the brain.
Following BMAA exposure on postnatal day (PND) 2 and/or PND 5 Dawson et al. observed reduced cerebellar weights in male and female rats and neuro-chemical changes in BMAA-exposed male rats, but not in exposed female rats. These changes, that are indicative of alterations in the development of the spinal cord and cerebellum, were accompanied by increased hind leg splay in male and female BMAA-exposed rats and a mild elevation in the blood pressure in female rats. Karlsson et al., reported that exposure to 600 mg BMAA/kg on PND 9 and 10 may lead to some mild disturbances in motor function and failure to habituate to a novel test environment and that early exposure may result in cognitive impairments in adulthood, but were unable to fully correlate the observed behavioral deficits to symptoms typically observed in ALS/PDC, AD and/or PD patients. Dobbing and Sands reported that the BGS, defined as the total brain weight gain as a percentage of the adult weight, peaks at PND 7 in rats, and not at PND 10, while Gottlieb reported that an initial rapid increase in dendrite density and brain complexity occurs between PND 0–6, as was also observed by Baloch et al., and Bockhorst et al., causing Semple et al. to suggest that developmental toxicity studies should, as a rule of thumb, be conducted on rats aged 4–7 days. Although Karlsson et al. have demonstrated the transplacental transfer of BMAA in rats, behavioral studies and neuro-pathology that support the toxicity of in utero BMAA exposure are lacking.
The observation that neonatal and adult rats respond differently to BMAA, together with the findings of epidemiological studies conducted by Garrutto et al., and Sabel et al., showing that exposure to environmental factors in utero or in the early stages of life, may be important for the development of ALS several years later, suggests that age of exposure might be the determining factor of BMAA neurotoxicity.
Reported deficits or behavioral abnormalities observed in BMAA-exposed animals or in other accepted animal models of AD and PD, have thus far failed to match all of the deficits that are commonly seen in patients with AD, PD, ALS. 6-OHDA results in impaired motor coordination and decreased locomotion of rodents in the open field test, but data on anxiety-like behavior, mood changes, visuo-spatial, long-term and working memory deficits, all symptoms frequently associated with PD, remain inconclusive. The MPTP mouse model fails to encompass the wide assortment of motor impairments seen in PD patients. Rotenone exposure does result in most PD-like motor symptoms and neuro-pathology, but seems to improve rather than impair spatial learning and memory abilities in treated mice. There also appears to be an intrinsic resistance of some rats to rotenone, and as few as 50% of treated animals within an experimental group exhibits neuro-degeneration and in the rats that do respond to rotenone, the time span during which the individual rats develop the symptoms, varies considerably. Without a suitable animal model for AD, PD and ALS/PDC it is impossible to investigate disease mechanisms or potential therapies, or conduct preclinical studies.
Knowledge of the effects of exposure to BMAA at different developmental stages remains limited and there is no report that compares the potential sex-dependent BMAA-induced alterations in behavioral, emotional and cognitive responses following in utero exposure, or exposure at critical stages of neonatal brain development. We therefore sought to investigate the effect, measured by standard rodent behavioral and cognitive response tests, of BMAA on male and female rats when exposed to the neurotoxin on gestational (G) day 14, PND 3, 4, 5, 6, 7 and 10 and from this potentially develop the first rodent model for ALS/PDC. These exposure times were selected to target specific developmental stages in the rodent brain so as to identify potential brain regions and/or processes involved in BMAA toxicity. BMAA exposure on G 14 aimed to target the developing brain just before the onset of formation of the dentate gyrus (G 18) and hippocampal regions (G 15.5). Postnatal BMAA exposure, conversely, was intended to target hippocampal and dentate gyrus neurogenesis which peaks at PND 3, 5 and 5, with 80% of cells being produced at this age and substantially decreases at the start of PND 7. Neurogenesis forms the basis for the normal structure and function of the adult brain and plays a critical role in the formation of hippocampal-dependent spatial learning and memory function later in life. At PND 0–2, the first distinct patches of dopamine fibers are distributed throughout the striatum after which dopaminergic innervation increases substantially throughout the developing brain Dopamine and serotonin modulate several aspects of neuronal development, including cell proliferation, migration and differentiation, and furthermore contribute to the development of pathways needed for movement, cognition and reward. Altered dopaminergic and serotonergic signaling during development can therefore produce long lasting changes that contribute to neuro-psychiatric and neuro-degenerative disorders’’ [16].
Uversky VN., “The etiologic basis of Parkinson’s disease (PD), the second most common age-related neuro-degenerative disorder, is unknown. Recent epidemiological and experimental studies indicate that exposure to environmental agents, including a number of agricultural chemicals, may contribute to the pathogenesis of this disorder. Animal models are important tools in experimental medical science for studying the pathogenesis and therapeutic intervention strategies of human diseases. Since many human disorders do not arise spontaneously in animals, characteristic functional changes have to be mimicked by neurotoxic agents. Agricultural chemicals, when administrated systemically, have been shown to reproduce specific features of PD in rodents, thus opening new routes for the development of animal models for this disorder. This study provides a detailed description of exiting models in which Parkinsonism is initiated via the exposure of animals to such agricultural chemicals as rotenone, paraquat, and maneb. Suggested neurotoxicity mechanisms of these chemicals are considered, and the major lessons learned from the analysis of pesticide-induced PD models are discussed” [17].
de Pedro N et al., “The development of new anti-neoplastic drugs is a key issue for cancer chemotherapy due to the reality that, most likely, certain cancer cells are resistant to current chemotherapy. The past 2 decades have witnessed tremendous advances in our understanding of the pathogenesis of cancer. These advances have allowed identification new targets as oncogenes, tumor supressor genes and the possible implication of the mitochondria. Annonaceous Acetogenins (ACGs) have been described as the most potent inhibitors of the respiratory chain because of their interaction with mitochondrial Complex I. To explore a possible application of natural products from Annonaceous plants to cancer treatment, we have selected four bis-tetrahydrofuranic ACGs, three from Annona cherimolia (cherimolin-1, motrilin and laherradurin) and one from Rollinia mucosa (rollinianstatin-1) in order to fully describe their mechanisms responsible within the cell. In this study, using a hepato-carcinoma cell line (HepG2) as a model, we showed that the bis-THF ACGs caused cell death through the induction of the apoptotic mitochondrial pathway. Their potency and behavior were compared with the classical mitochondrial respiratory chain Complex I inhibitor rotenone in every apoptotic pathway step” [18].
Degli Esposti M et al., “Natural products from the plants of the family Annonaceae, collectively called Annonaceous acetogenins, are very potent inhibitors of the NADH-ubiquinone reductase (Complex I) activity of mammalian mitochondria. The properties of five of such acetogenins are compared with those of rotenone and piericidin, classical potent inhibitors of Complex I. Rolliniastatin-1 and rolliniastatin-2 are more powerful than piericidin in terms of both their inhibitory constant and the protein-dependence of their titre in bovine submitochondrial particles. These acetogenins could be considered therefore the most potent inhibitors of mammalian Complex I. Squamocin and otivarin also have an inhibitory constant lower than that of piericidin, but display a larger protein-dependence of the titre. Squamocin and otivarin, contrary to the other acetogenins, behave qualitatively like rotenone. Rolliniastatin-2 shows unique properties as its interaction, although mutually exclusive to that of piericidin, appears to be mutually non-exclusive to that of rotenone. It is the first time that a potent inhibitor of Complex I is found not to overlap the active site of rotenone” [19].
Segura Aguilar J et al., “Neurotoxins, in the general sense, represent novel chemical structures which when administered in vivo or in vitro, are capable of producing neuronal damage or neuro-degeneration--with some degree of specificity relating to neuronal phenotype or populations of neurons with specific characteristics (i.e., receptor type, ion channel type, astrocyte-dependence, etc.). The broader term ‘neurotoxin’ includes this categorization but extends the term to include intra- or extracellular mediators involved in the neurodegenerative event, including necrotic and apoptotic factors. As it is recognized that astrocytes are essential supportive satellite cells for neurons, and because damage to these cells ultimately affects neuronal function, the term ‘neurotoxin’ might reasonably be extended to include those chemical species which also adversely affect astrocytes. This review is intended to highlight developments that have occurred in the field of ‘neuro-toxins’ during the past 5 years, including MPTP/MPP+, 6-hydroxydopamine (6-OHDA), methamphetamine; salsolinol; leukoaminochrome-o-semiquinone; rotenone; iron; paraquat; HPP+; veratridine; soman; glutamate; kainate; 3-nitropropionic acid; peroxynitrite anion; and metals (copper, manganese, lead, mercury). Neuro-toxins represent tools to help elucidate intra- and extra-cellular processes involved in neuronal necrosis and apoptosis, so that drugs can be developed towards targets that interrupt the processes leading towards neuronal death” [20].
Caparros-Lefebvre D et al., “Over the last 60 years an abnormally high prevalence of atypical Parkinsonism has been reported in 5 different geographic isolates. It was first described on Guam, later in New Guinea and in the Kii peninsula, on Guadeloupe, and in New Caledonia. We investigated the phenotype of atypical Parkinsonism in 3 of these foci and observed several similarities with dementia in most and amyotrophic lateral sclerosis in some. This disappearance of this disease in two places--Guam and New Guinea--suggested an environmental origin which has not been clarified before the disease ended. The exposure to annonaceae acetogenins and/or rotenone has been documented in four of these places, and experimental studies in animals demonstrated annonaceae acetogenins neuro-toxicity, which is similar to rotenone neuro-toxicity. Simultaneous exposure to acetogenins and rotenone could produce a synergistic toxicity on neurons” [21].
Khandare AL et al., “Lathyrism is a disease caused by excessive consumption of grass pea, Lathyrus sativus especially under conditions of severe drought. Grass pea contains 3-N-oxalyl-L-2, 3-diaminopropanoic acid (β-ODAP) a putative neuro-toxin which acts through excitatory mechanism causing Neurolathyrism. Due to awareness of the disease, availability of food and levels of consumption of L. sativus there is reduction in lathyrism cases where higher consumption of L. sativus is reported in India. The present study was undertaken with the objective to assess the current scenario of consumption of L. sativus, incidence of cases of lathyrism, β-ODAP, protein and amino acids content in L. sativus pulse collected from 3 districts of Chattisgarh state. For this purpose, a total of 17,755 (13,129 rural and 4626 urban) individuals from 151 villages and 60 wards from urban area were covered for clinical examination. Out of total 5769 households (HHs) covered during the survey, 1602 HHs were cultivators, 1791 HHs non-cultivators and 2376 agricultural and other labourers. A one day 24-hour re-call diet survey was carried out in 5758 HHs (4549 rural and 1209 urban). 360 split grass pea (SGP) samples were collected to estimate β-ODAP, protein and amino acids content. Results of the study revealed that an average consumption of SGP was 20.9 gm/CU/day in Bilaspur and no consumption was reported among urban population of Raipur. Only nine old cases of lathyrism were found during the study. The mean β-ODAP content in SGP was 0.63 ± 0.14, 0.65 ± 0.13 and 0.65 ± 0.14 gm/100 gm, whereas the protein content was 27.0 ± 2.39, 27.0 ± 1.99 and 26.7 ± 1.90 gm/100 gm in samples collected from Bilaspur, Durg and Raipur districts respectively. Arginine content was high in SGP and sulphur containing amino acids (cysteine and methionine) were less than other amino acids. In conclusion, the consumption of SGP was lower in these 3 districts with lower β-ODAP content than earlier reports, thus the lower prevalence of lathyrism in the districts surveyed” [22].
Mohamed Salama et al., “While use of pesticides has been suggested to be partially responsible for PD in rural areas, this is not correlated to disease prevalence, as the odds ratio for farming itself cannot be accounted for by pesticide exposure alone. Associated with rural living, humans have a strong relation with the surrounding environment, and both individual exposure (e.g. occupation) or group exposure (e.g. drinking well water) are present. Exposure to environmental agents may contribute to the onset or progression of PD.
The success of new environmental toxins to develop mitochondrial complex I inhibition and degeneration of dopaminergic neurons in vitro, coupled with epidemiological data suggesting natural environmental toxin involvement in Parkinsonism pathogenesis would invite us to think of these natural toxins as new candidates for developing models.
The choice of certain natural toxins for PD modeling needs some guidelines as supposed by Shaw and Höglinger. Based on many works dealing with natural toxins some properties must be fulfilled to make them suitable candidates for toxic models. These characteristics include:
The agent must be of natural origin.
The agent must be available worldwide to contribute to the wide prevalence of PD in the whole world.
The agent must recapitulate PD pathology in experimental animals.
The following natural agents represent promising candidates for application in experimental PD models. Although they do not show the criteria of an ideal PD model, we must put in consideration the fact that they need further research regarding the dosing regimen and species vulnerability before comparing their value to the classical toxins of PD models.
The Cycadaceae (cycad) family contains about 45 primitive species of seed-bearing plants that probably dominated the world’s vegetation during the Jurassic and early Cretaceous periods. Cycads, like all plants, produce a variety of secondary substances. The most important of such allelochemicals are, in addition to dimeric flavones, the nitrogen-containing methylazoglucosides cycasin, macrozamin and several neocycasins. There is a minor cycad toxin which is an unusual, nonprotein amino acid, a derivative of alanine (beta-methylamino alanine [BMAA]), which in higher concentrations has been found to be neurotoxic to mammals and chickens.

Figure 15: Grass pea (Lathyrus sativus).
The linkage between cycads and neurodegenerative diseases began in 1945. In that year Zimmerman reported cases of motor neuron disease among Chamorros, the local citizens of Guam. Later, it was confirmed that the disease was clinically and pathologically similar to amyotrophic lateral sclerosis (ALS), although it was familial and confined to local Chamorros (it was 100 times more prevalent than elsewhere in the world).
Another atypical Parkinson-like syndrome was identified with onset in middle life and accompanied by mental slowness. This syndrome was noticed in the same families who developed the ALS syndrome. In 1961, Hirano and colleagues introduced the term Parkinsonism–dementia complex (PDC) of Guam, and they also noted the widespread presence of Alzheimer-type neurofibrillary tangles (NFTs) in those with the syndrome. It seemed that the ALS and PDC syndromes represented the spectrum of a single disease. This hypothesis was based on the observation of NFTs in both clinical entities.
2 additional links between the Guam syndrome and other neurodegenerative diseases appeared. The first is the transactive response (TAR) DNA-binding protein 43 (TDP-43) and the second is the leucine-rich repeat kinase 2 (Lrrk2). TDP-43 functions as a transcriptional repressor and splicing regulator where it is present in ubiquitin-positive, tau and α-synuclein negative inclusions in frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U) and in ALS, it has been shown to occur in some cases of Lewy body disorders alone or in association with AD. Although the role of TDP-43 in these proteinopathies is unknown, it may provide a common pathologic link.
LRRK-2 (which is strongly linked to autosomal dominant Parkinson disease) may be central to the pathogenesis of several major neurodegenerative disorders associated with Parkinsonism. Pathologic studies of both ALS/PDC and nonsymptomatic Chamorro cases reveal that they are more prone to NFT development. Immunohistochemical studies reveal that the tau isoform distribution is similar to AD; the cortical laminar distribution is similar to progressive supranuclear palsy (PSP). In these lesions, immunostaining with antibodies to TDP-43 and LRRK-2 shows a commonality with PD, ALS, and some other neurodegenerative disorders.
Failure to find any genetic abnormalities in Guam cases led scientists to think about an environmental component and more specifically Cycas micronesica which is the source of flour of fadang, a traditional food in this locality.
The first incriminated toxic component of C. micronesica was the nitrogen-containing methylazoglucosides cycasin. In rat neuronal cultures, the aglycone of cycasin methylazoxymethanol (MAM) can damage neuronal DNA, disrupt glutamate transmission, and promote the accumulation of tau protein and neuronal degeneration. Failure to induce neuropathology in experimental animals using cycasin excluded it from being the causative toxin.
The possible role of BMAA in ALS/PDC replaced the old cycasin theory; it was not before 1987 when Spencer and colleagues managed to show the capacity of BMAA to induce pathological changes in primates’ brain. The interest toward BMAA as a putative toxic agent damaging DAergic cells has grown considerably due to the discovery that this toxin is not only present in the Guamanian islands but appears to be ubiquitous throughout the world. This natural amino acid deposits in human tissue proteins. This protein binding represents a source of chronic exposure through protein catabolism. BMAA is distributed worldwide with the chance of frequent repetitive exposures.
Studies properly quantified the concentration of BMAA in postmortem brain tissues of patients. The deleterious mechanisms of the toxin could underlie neurological conditions such as ALS–PDC and, potentially, idiopathic PD. Some studies have investigated the effects of BMAA on nigrostriatal DAergic neurons either indirectly (by measuring the ability of these cells to uptake or release dopamine) or directly (through studying the effects of BMAA on single DAergic neurons of the SNpc in an in vitro slice preparation). It was found that brief BMAA applications, at a concentration similar to that found in ALS–PDC brains, induced reversible and reproducible membrane depolarizations/inward currents, firing increase, and Ca2+ accumulation. Prolonged exposures (12 min) caused functional impairment, cellular shrinkage, calcium overload, cyt-c release, and ROS production that did not return to control values for up to 20 min or several hours after BMAA washout.
Cucchiaroni et al., the action of BMAA may be due to activation of TRPC-like channels, through mGluR1. Interestingly, GABAergic SNpc cells respond to BMAA with an AMPA-mediated inward current that is not associated with an intracellular calcium increase. BMAA has been previously reported to activate mGluRs in hippocampal, striatal, and cerebellar neurons. Alternatively, it predominantly activates NMDA receptors in cortical neurons and AMPA/kainate receptors in spinal motoneurons.
The BMAA hypothesis, faced criticism because of the high doses used in the experiment which cannot be true to real life. This criticism led to fading of the BMAA theory for a while. The enthusiasm towards BMAA was regained with recent findings of the possible exposure to high doses (in contrast to what was expected before) through the bound fraction of the toxin rather than the free amino acid, the biomagnification of BMAA exposure through eating animals previously fed on the cycad plants, e.g. flying fox, and the transfer of this cyanotoxin through aquatic ecosystems suggesting further human exposure.
BMAA as an inducer of PD faces a lot of debate; there is no detected animal model for such a toxin; second, it lacks specificity towards SN. In other words, BMAA cannot be considered a specific PD-inducing toxin. The previous research points out that cycads are not an attractive research tool in the field of PD modeling. a recent finding of the possible role of cycads was based on another constituent (sterol glucosides).
Recently a new direction rose proposing a new component in cycads as the agent responsible for neurodegeneration. These sterol b-D-glucosides have been isolated by Schulz and colleagues, who found that these glucosides induce the excitotoxic release of glutamate.
Shen and colleagues used washed cycad seeds to make flour pellets (the washed seeds are devoid of BMAA) and fed them to outbred Sprague-Dawley rats the experiments were performed with the intention of developing a model of ALS–PDC. The rats showed Parkinsonism pathology without ALS syndrome. Also the cycad-fed rats showed the gradual development of multiple PD motor abnormalities after 2–3 months of feeding, including spontaneous unilateral rotation, shuffling gait, and stereotypy. Histological and biochemical examination of brains from cycad-fed rats revealed an initial decrease in the levels of dopamine and its metabolites in the striatum, followed by neurodegeneration of dopaminergic cell bodies in the SNc. Alpha-synuclein and ubiquitin aggregates were found in the DAergic neurons of the SNc and neurites in the striatum (STR). α-synuclein aggregates were found in the neurons of the locus coeruleus and cingulate cortex. In an organotypic slice culture of the rat SN and the striatum, an organic extract of cycad causes a selective loss of dopamine neurons and α-synuclein aggregates in the SN.
Unlike genetic models or toxicant-based end-stage models of Parkinsonism that may have a great deal of phenotypic similarity among animals, there is considerable variability in the response of individual animals to cycad ingestion. One of the strengths of this cycad-fed rat model is that it recapitulates the variability and progression of symptoms and neuropathology seen in human Parkinsonism. This model also allows examination of the early stages of the disorder based on the chronic progressive nature.
This new trend of using the seeds without attempting to extract a single toxic component may prove useful. In fact, it seems that this approach is more representative of real life exposure. Despite the debate about which toxic component in cycads is responsible for neurodegeneration, they represent a possible new substance for PD modeling with proven capacity to induce progressive and chronic neurodegeneration in a fashion similar to reality. To sum up, we can see that cycad offers a possible natural toxin candidate. It has been linked epidemiologically to a neurodegenerative condition with pathological similarity to PD: cycads are available worldwide and their use in in vivo studies could recapitulate the pathological features of PD.
Although cycad seeds have been linked to ALS/PDC syndrome, the recent findings of Shen and colleagues introduce them as promising tool for modeling PD experimentally.
Epoximicin is a natural proteasome inhibitor (PI). A product of actinomycetes bacteria which live in moist soil. When the bacteria release this toxic metabolite it can pass into the underground water store providing a route for human exposure.
PIs are small, bioavailable molecules that attack the 20S proteasomal subunit, the primary site responsible for the cleavage of peptide bonds. By covalently binding to the 20S subunit, PIs successfully block proteolysis and degradation of proteins. Defects in the ubiquitin–proteasome system (UPS) and degradation of unwanted proteins are considered a common feature in PD. in hereditary forms of PD, mutations in the UPS (parkin and ubiquitin C-terminal hydrolase L1) are responsible for the development of the disease. Mayer used the Cre-recombinase/loxP genetic approach to ablate the proteasomal Psmc1 ATPase gene and deplete 26S proteasomes in neurons in different regions of the brain to mimic neurodegeneration. This approach generates dopaminergic neurodegeneration accompanied by Lewy pathology. Based on the previous data, proteasome inhibition can be used to induce PD.
The idea of using epoximicin as a systemic PI in PD modeling was presented by McNaught et al., who showed that systemic exposure of rats to epoximicin can induce a model of PD. After a latency of 1–2 weeks, treated animals develop a gradually progressive, L-dopa/apomorphine-responsive, Parkinsonian syndrome with a decrease in striatal 11C-CFT binding on positron emission tomography (PET). Pathologically, there is neurodegeneration in the SNc, as well as in the locus coeruleus (LC), dorsal motor nucleus of the vagus (DMN), and nucleus basalis of Meynert (NMB), a pattern of neuronal loss similar to that found in PD. neurodegeneration is accompanied by intracytoplasmic Lewy body-like inclusions that stain positively for α-synuclein, ubiquitin, and other proteins. Thus, PIs can induce a Parkinsonian syndrome in rats that more closely recapitulates the features of the illness in humans than other models.
The same model has not been reproducible in many trials in rodents in spite of using the same doses as used initially. This raised concerns about the possible application of epoximicin in animal models of PD. Matsui et al, demonstrated that the administration of this natural PI to medaka fish via the cerebrospinal fluid (CSF) induces PD-like symptoms in the form of reduced spontaneous movement. Fish treated with epoximicin developed inclusion bodies similar to Lewy bodies throughout the central nervous system, which are associated with PD. Treatment with epoximicin also induced selective loss of dopaminergic and noradrenergic neurons. This model replicated the cardinal signs of PD in the form of locomotor dysfunction, selective dopaminergic cell loss, and inclusion body formation. The reason for this controversy between models is unknown, but in medaka the TH-positive neurons have relatively large cell bodies with well-developed fibers. These large neurons may require more optimal conditions for survival and this may explain the increased vulnerability of these cells. The problems with animal models of epoximicin can be solved by increasing the dose to overcome the lesser vulnerability of animal neurons compared with the medaka fish.
These findings demonstrate that epoximicin can be considered to be an effective dopaminergic neurotoxin that may offer a good model for PD, as it is widely distributed, linked epidemiologically to PD, and can re-induce the pathological features of PD experimentally.
The epidemic of postencephalic Parkinsonism was first described by von Economo in 1917. This gave rise to the belief that PD might be associated with an infectious agent such as a virus, more infectious agents such as Borrelia burgdorferi, Mycoplasma pneumoniae and Helicobacter pylori have been postulated as possible inducing agents for Parkinsonism.
There is a well-documented animal model for PD in mice, using the GUH-2 strain of Nocardia asteroides. N. asteroides is a bacterial strain found worldwide that is commonly isolated from soil and aquatic sources.
It is a Gram + aerobic bacterium that primarily affects the lung and skin. Pathogenic strains can cause a variety of pulmonary, skin, systemic and brain lesions in immunocompromised or normal humans. N. asteroides (GUH-2) is neuroinvasive after tail vein injection into mice.
The first experimental trial of N. asteroides was conducted by Kohbata and Beaman who through multiple injections of this pathogen were able to induce a PD-like condition in mice which was later confirmed by postmortem pathological examination. They found that after administration of a nonlethal dose of log phase cells of GUH-2 (<200 colony forming units reaching the brain), Nocardiae invaded through the capillary endothelium, crossed the basal lamina, and then grew perivascularly within the brain parenchyma without associated inflammatory responses. These bacteria grew primarily within neurons, along axons, and in astroglia. Approximately 10 days after infection, mice developed one or more features of neurological impairment. Later on, the headshaking, stooped posture, and bradykinesia tended to persist for the life of the mouse. Administration of levodopa (20 mg/kg i.p.) caused transient reversal of head-shaking, tremors, and hypoactivity. These abnormal movements were confirmed by histopathological analysis of the brains from mice. Histopathological findings showed decrease of neurons in the substantia nigra, a loss of Nissl substance in the remaining neurons, and diminished immunostaining for tyrosine hydroxylase in the substantia nigra pars compacta. The previous data suggest a possible dopaminergic involvement in the mice inoculated with GUH-2. Interesting finding in this model was the selective reduction in neostriatal DA concentrations at 1 week after GUH-2 inoculation, without a change in NE or 5HT concentrations. The selective changes for neostriatal metabolism at 1 week suggest that GUH-2 has specificity for nigral neurons engaged in dopamine synthesis. A significant finding in the case of idiopathic PD is the Lewy body, although no animal model of Parkinsonism has led to their generation. In the case of GUH-2-induced models, hyaline-like intraneuronal inclusions were found in the murine brain, which is considered a major strength of this type of model with regards to its relation to the progression of disease.
Based on many studies, we can say that Nocardia induce neurodegeneration in vivo and in vitro similar to that seen with MPTP, supporting this model for the study of Parkinsonism.another study used N. otitidiscaviarum (GAM-5) which is isolated from a patient with an actinomycetoma. GAM-5 produced signs similar to PD following intravenous administration to NMRI mice. Motor signs included head tremor, akinesia/bradykinesia, flexed posture, deviation of the head, trunk flexion, and postural instability in a manner similar to N. asteroides. In both models (GAM-5 and GUH-2), the loss of bacteria in the brain coincided with the appearance of the motor impairments described above.
Why the mice developed signs of Parkinsonism after the apparent elimination of the bacteria from the brain is still unknown. A possible explanation could be the release of neurotoxic substances from the nocardial cells as they were killed and cleared from the brain.
A final strength supporting the use of these bacteria in PD models is the intracytoplasmic hyaline-like inclusion bodies in neurons, mostly in the substantia nigra, that resemble Lewy bodies. Whether or not the chemical composition of these intracytoplasmic hyaline-like inclusion bodies is the same as that found in PD remains to be determined. Nocardia-induced PD animal models represents a new, different, and promising approach based on the previous findings.
Other natural agents have been suggested as possible substitutes to the classical PD-inducing agents. Some are of bacterial origin, e.g. Streptomyces venezuelae, and others are of plant origin, e.g. annonacin. On Guadeloupe Island, the consumption of Annona muricata has been linked to sporadic taupathy.
In A. muricata (annonacin) can easily cross biological membranes, owing to its high lipophilicity, so is capable of entering nerve cells. After chronic exposure in rats, neuronal cell loss and gliosis were observed in the brain stem and basal ganglia.
The aggregation of the microtubule-associated protein (tau protein) characterizes several neurodegenerative diseases known collectively as tauopathies. While PD, is the best known of the α-synucleinopathies, tau pathology is also seen in many PD cases. Staining of some Lewy bodies with multiple tau antibodies has been reported, suggesting cellular colocalization of these two pathologies. Höllerhage and colleagues found that lipophilic complex I inhibitors of natural or synthetic origin can reproduce in vitro tauopathies. Exposure to annonacin led to phosphorylation and somatodendritic redistribution of tau in a manner similar to MPTP effects. Epidemiological and bioanalytical studies should be undertaken to verify this hypothesis. In vivo experiments are needed to combine these findings with the data on absorption, distribution, metabolism, and elimination (ADME) which are still lacking.
Another agent suggested by Panneton and colleagues was the dopamine metabolite DOPAL. They demonstrated that injections of DOPAL selectively kill SN DA neurons and trigger a behavior consistent with other PD animal models. This study supports the ‘catecholaldehyde hypothesis’ as an important link for the etiology of sporadic PD” [23].
Virginia Bozzoni et al., “Cyanotoxins Exposure to the neurotoxic non-protein amino acid β-N-methylamino-L-alanine (BMAA) may play a role in neurodegenerative processes.
BMAA is produced across the cyanobacterial order and it has been detected in several aquatic and terrestrial environments worldwide, suggesting that it is ubiquitous.
Cyanobacteria produce several cyanotoxins, divided into neurotropic (e.g. BMAA) and hepatotropic, such as cycasin, whose carcinogenic potential is well documented. A considerable BMAA concentration is present in cycad seeds (Cycas micronesica), used by the Guam indigenous population to produce flour, even though Duncan and coworkers report that it is largely removed during the washing of the seeds: these authors analyzed 30 flour samples and revealed that most of the neurotoxic amino acid (almost 87%) was removed during processing.
The hypothesis that BMAA may have a role in neurodegenerative diseases was initially based on reports of elevated rates of ALS, Parkinson’s disease and dementia in the island of Guam, a finding which gave rise to the term Western Pacific ALS-Parkinson Dementia Complex (ALS-PDC). A remarkable incidence of these neurodegenerative diseases was first noticed there in 1904, and in 1945 the ALS-PDC incidence rate in Guam was 50 to 100 times higher than overall world incidence. Recent studies have shown a decreasing incidence rate in the area in the last four decades, even though it remains 3 times higher than the overall one.
In vivo experiments were developed: in 1987, Spencer and collaborators conducted one in which they fed BMAA (100 to 250 mg/kg) to macaques for 12 weeks; they observed numerous neurological symptoms, which suggested the presence of an active neurodegenerative process.
In spite of these epidemiological data, this hypothesis was initially challenged because the amount of BMAA contained in the dietary flour of the Guam population was considered insufficient to trigger neurodegeneration.
The role of BMAA as a risk factor for ALS has since been reconsidered on the strength of the finding that various animals consuming cycad seeds, such as flying foxes, pigs and deer, contribute to biomagnification of BMAA through the food chain in Guam.
The BMAA concentration at autopsy in the nervous tissues of deceased Chamorro indigenous people affected by ALS/PDC, together with its absence in indigenous people who died for other reasons, strengthens the hypothesis of a role for BMAA in ALS/PDC in Guam.
Increased brain levels of BMAA were later confirmed by Pablo et al. (2009), who also found similar BMAA levels in the brains of ALS patient in Florida, but not in the brains of healthy subjects.
Pablo et al., evaluated BMAA in neuroproteins of post-mortem brain tissues of 13 ALS patients, 12 Alzheimer’s disease patients, eight Huntington’s disease patients and 12 controls who died of non-neurological causes. BMAA quantification was performed using the fluorescent high performance liquid chromatography method previously used to detect BMAA in ALS/PDC patients in Guam. Tandem mass spectrometric analysis was carried out to confirm the identification of BMAA in neurological specimens. The concentrations of BMAA were below what had been reported previously in Chamarro ALS/PDC patients, suggesting that BMAA can, in any case, considered a risk factor.
The latest in vivo studies confirm that BMAA can be incorporated into nerve cell proteins, causing misfolding, aggregates and cell death.
In an interesting review, Chui et al., summarized the principal mechanisms of action of BMAA: BMAA crosses the blood-brain barrier and, after reacting with bicarbonate ions, forms β-carbamate, which can bind to glutamate receptors and stimulate noradrenaline release. This amino acid enhances oxidative stress through glutathione depletion and stimulates cytochrome-c release from the mitochondria, inducing cellular apoptosis.
Dunlop et al., state that “motor neurons, as they are post-mitotic cells, are particularly vulnerable to aggregated protein damage since they are unable to reduce it by distributing [aggregates] among daughter cells”. They state that misfolded proteins, resulting from misincorporation of non-protein amino acids, can be transmitted within the brain through a prion-type mechanism; this may trigger the neurodegenerative cascade. In the same study, Dunlop et al., analyzed the effects of BMAA exposure in cultured cells (fibroblasts, human neuroblastoma cell line and human endothelial cells), focusing in particular on the incorporation of BMAA into human proteins. They demonstrated that BMAA is incorporated by a protein synthesis-dependent mechanism, since the incubation of human cells with BMAA and a protein synthesis inhibitor significantly reduced BMAA incorporation. It was also found that incorporation of BMAA into cell proteins was inhibited in the presence of L-serine in a concentration-dependent manner, while the same effect was not observed with D-serine. The incorporation of BMAA into proteins induced apoptosis in neuroblastoma cells in vitro, an effect previously reported with other non-protein amino acids. Auto fluorescence developed in perinuclear and cytosolic regions of the cells incubated with 300 mM BMAA, suggesting the presence of protein aggregation. This phenomenon was prevented by co-incubation with an equimolar concentration of L-serine.
The authors of a recent retrospective study evaluated BMAA exposure in ALS cases in whom the disease was diagnosed between 1994 and 2009 in the ALS center of Montpellier; their study included cluster analysis and evaluation of BMAA sources in this area. One significant ALS cluster was observed in the area surrounding the Thau lagoon, one of the main areas of shellfish production and consumption on the French Mediterranean coast: 68 cases were detected there (as opposed to 33.7 expected cases), leading to a standardized incidence ratio (SIR) of 2.02 and RR of 2.24 (p=0.0024). The BMAA concentration in mussels and oysters was also measured and it was found that the level was higher during the summer, when cyanobacteria, considered the source of this amino acid, bloom.
BMAA: conclusions: BMAA remains a possible, non-proven risk factor for ALS/PDC in Guam. Although there is some in vivo evidence that validates its neurotoxic properties, epidemiological evidence is substantially lacking and further investigations are necessary to prove the role of BMAA in sporadic ALS throughout the world.”
Pesticides: Strong evidence indicates that pesticides play a role in ALS. This, in addition to the involvement of pesticides in other neurodegenerative diseases such as Parkinson’s disease, leads to the conclusion that they are a reliable risk factor for neurodegeneration” [24].
Heavy metals
The role of exposure to heavy metals, such as lead, selenium, mercury, cadmium and iron, as a risk factor for ALS has long been studied, and the results produced are contradictory. The potential role of several heavy metals as contributors to molecular mechanisms leading to motor neuron degeneration has been widely explored but only partially characterized (Trojsi et al., 2013).
A recent systematic review analyzed 50 studies dealing with the relationship between metals and ALS and found only three studies that suggested an association. One study reported significantly higher ALS risk for individuals exposed to selenium (risk estimate 5.72, 95% CI 1.46–15.57). Another study concerning the effect of lead, mercury, aluminum, cadmium, chromium and manganese revealed no significant associations (Sutedja et al., 2009).
The hypothesis that host genetic background can modify the response to xenobiotics has long been postulated. Polymorphisms have been found in the genes encoding for delta-aminolevulinic acid dehydratase (ALAD) and for vitamin D receptor (VDR), while an impaired ability to detoxify pesticides and heavy metals, due to polymorphisms on metallotionein (MT), transcription factor (MTF-1) and glutathione synthetase (GSS) genes, was detected in several ALS patients (Morahan et al., 2007).
With regard to heavy metals, the ALAD and VDR genes may be the major ones involved in determining genetic susceptibility.
ALAD (9q34) encodes for an enzyme implicated in heme group synthesis in erythrocytes and is the major Pb-binding protein inside these cells. A single nucleotide mutation, located in position 177 of the ALAD gene-coding region (K59N), is able to enhance the protein’s affinity for lead and can alter the consequences of exposure to it.
Although a positive correlation seemed to emerge in the first studies (Kamel et al., 2003), a later paper (Fang et al., 2010) failed to detect the same correlation.
Vitamin D can influence lead absorption and distribution. Even though various studies have attempted to validate this assumption, the evidence in the literature does not confirm the involvement of the VDR gene (12q) in a genetic susceptibility to lead in ALS (Kamel et al., 2003).
Lengthy and in-depth study of the relationship between Pb exposure and genetic susceptibility has given conflicting and inconclusive results. There has been considerable interest in the possible role of selenium, particularly in South Dakota and in Northern Italy (Reggio Emilia) on account of the presence of seleniferous drinking water in these areas (Trojsi et al., 2013). Evidence based on animal and cell culture studies confirmed that mercury is involved in ALS pathogenesis, epidemiological case-control studies showed no relationship between Hg exposure and ALS incidence.
There exists in vivo but not epidemiological evidence of neurotoxic properties of cadmium and iron in the human population [24].
Francisco Pan-Montojo et al., “Neurodegenerative diseases are characterized by a progressive dysfunction of the nervous system. Often associated with atrophy of the affected central or peripheral nervous structures, they include diseases such as Parkinson’s Disease (PD), Alzheimer’s Disease and other dementias, Genetic Brain Disorders, Amyotrophic Lateral Sclerosis (ALS or Lou Gehrig’s Disease), Huntington’s Disease, Prion Diseases, and others. The prevalence of neurodegenerative diseases has increased over the last years. This has had a major impact both on patients and their families and has exponentially increased the medical bill by hundreds of billions of Euros. Therefore, understanding the role of environmental and genetic factors in the pathogenesis of PD is crucial to develop preventive strategies. While some authors believe that PD is mainly genetic and that the aging of the society is the principal cause for this increase, different studies suggest that PD may be due to an increased exposure to environmental toxins. In this article we review epidemiological, sociological and experimental studies to determine which hypothesis is more plausible. Our conclusion is that, at least in idiopathic PD (iPD), the exposure to toxic environmental substances could play an important role in its aetiology”.
“PD affects around 1% of the general population a rate that increases with age reaching up to 5% of the 80 years old population. Its annual incidence is between 16 and 19 per 100,000 per year. It affects all races equally, with slight male predomination. In Europe PD prevalence is 1,6/100 inhabitants, with a wide variability between different countries. Although multiple genetic forms of the disease have been observed, they account for less than 10-15% of the total cases and their importance vary between different regions. If iatrogenic and vascular parkinsonisms are included together with complex degenerative diseases and atypical parkinsonisms, the total prevalence increases to 2,3/100. During the twentieth century, the general incidence of PD increased 1.63 times. This increase occurred mostly during the first half until 1980 and affected developed countries. Similarly, other neurodegenerative diseases have dramatically increased in some regions and decreased in others. Interestingly, this increase is most evident in developed countries and within these countries in those regions using agrochemical compounds. Some studies claim that such an increase is due to the aging of the population, associated with mitochondrial dysfunction and a reduced protective response to oxidative stress, other studies point to the interaction between environmental toxins and diverse genetic backgrounds as the main triggers of PD. In this work, we show evidence supporting these two different hypotheses in relation with the most recent findings on PD pathophysiology with special emphasis on iPD.
The English physician James Parkinson in his work “An essay on the Shaking Palsy” described PD for the first time in 1817. This correlates well with the beginning of the industrial and chemical revolution in Europe during the late 18th century and the 19th century. Only the Ayuverda (the medical system practiced in India around 5000 years before Christ) and the first Chinese manuscript on medicine, Nei Ping, written 2500 years ago, describe some of the symptoms observed in PD and potential treatments. Apart from this reference to PD-related symptoms, no other physician in any of the occidental countries had previously described the complex amalgam of symptoms typical of this disorder. This suggests that either these different symptoms had always been misdiagnosed as separate entities throughout history, not recognized as part of a syndrome, or the prevalence of PD until the beginning of the 19th century had been extremely low. While possible, the first hypothesis seems quite improbable to us. Many PD symptoms are quite striking and would have been described and published before Parkinson’s assay. Therefore, we believe that a dramatic increase of PD cases occurred in parallel to the industrial revolution. In this case, the question is, why?
Some authors have related the increase of PD incidence to the aging of the population. Aging has been associated to the impairment of the antioxidant body system and mitochondrial function. It is known that age-related non-genetic PD appears between the 5th and 6th decade of life. Logically, an increase in life expectancy would lead to an increased incidence and prevalence of PD. in the last 1000 years life expectancy at 15 years of age (i.e. discounting birth and child mortality) has always been above these values. Interestingly, a general increase in PD incidence was observed during the first half of the twentieth century from 11,4/100.000 inhabitants between 1935 and 1944 to 18,2/100.000 inhabitants in both young and old populations between 1967 and 1979. Since then it has remained more or less constant due to a compensation effect. There has been a decrease in the incidence rate in the population under 69 years of age and an increase in the population over 70 years. These data suggests that the increase in PD incidence cannot be due to the aging of the population alone.
The appearance of iPD has been related to industrial and rural environments due to the higher exposure to environmental toxins. Natural pesticides have been used since more than 5000 thousand years. The first known pesticide was elemental sulfur dusting used about 4,500 years ago. Also, the Rig Veda, which is about 4,000 years old, mentions the use of poisonous plants for pest control. it was not until the 15th century that toxic chemicals such as arsenic, mercury, and lead were applied to crops to kill pests. Later, nicotine sulfate was extracted from tobacco leaves for use as an insecticide in the 17th century. Only in the 19th century two pesticides related to PD, pyrethrum, derived from chrysanthemums, and rotenone, which is derived from the roots of tropical vegetables, started to be used.
Other studies have broadened the link between PD and other environmental factors including drinking well water, rural living, farming, diet and exposure to agricultural chemicals. The environmental contribution to PD’s pathophysiology has been also analyzed. Farm and industrial compounds seem to increase the risk of Parkinsonism. Farm activity is associated with agriculture and exposure to pesticides. Organochlored pesticides were identified as a risk factor in a German case-control study and another study with similar conclusions was conducted with dithiocarbamates. Levels of organochlorines have been found to be elevated in the brains of persons with iPD. A study of French elderly individuals found an association between past occupational exposures to pesticides, low cognitive performance, and increased risk of developing Alzheimer’s disease or iPD. In a study, Tanner and colleagues have tried to identify a common characteristic of those pesticides that present a higher correlation to the appearance of PD. They conclude that pesticides inhibiting the mitochondrial Complex I and increasing oxidative stress are more prone to induce iPD upon exposure. Together with pesticides and herbicides it has been observed that some xenobiotics like annonacin induce Parkinsonian symptoms in humans and a loss of nigrostriatal neurons in animals.
iPD has also been linked to the exposure to different metals and industrial compounds. Many studies performed in the 90’s identified manganese, lead, copper, iron, zinc, aluminium or amalgam (reviewed in). Higher incidence of iPD has been reported in manganese miners. It was shown that manganese, a component of various pesticides, also reproduces parkinsonian symptoms after long and chronic exposures (between 6 months and 16 years). Also, concern was raised that widespread introduction of the manganese-containing fuel additive methylcyclopentadienyl manganese tricarbonyl to the U.S. gasoline supply may increase population exposure to manganese and thus increase risk of Parkinsonism in sensitive populations. A more recent study has also shown a positive correlation between ß-Hexachlorocyclohexane blood levels and iPD.
The positive relation between exposure to environmental toxins and neurodegenerative diseases is not limited to PD. For example, the Chamorros population of Guam and Rota in the western Pacific has an unusually high prevalence of motor neuron disease, a syndrome that includes amyotrophic lateral sclerosis, Parkinsonism, and progressive dementia. It was proposed that this syndrome of parkinsonian dementia is related to the consumption of flour made from cycad seeds or to inhalation of pollen from cycad plants. Later findings suggest that a neurotoxic non-protein amino acid, beta-methylamino-L-alanine synthesized by a symbiotic cyanobacterium highly present on cycad seeds and pollen is actually responsible for this effect [25].
Rafael H et al., “To date all researchers conclude that the etiology of Amyotrophic lateral sclerosis (ALS) is not known. On the contrary, since August 2009, we believe that disease is of ischemic origin in the anterior surface of the medulla oblongata.
We present our surgical experience into 45 patients with ALS (bulbar form in 36 cases and spinal form in 9). Preoperative MRI scans revealed microinfarcts in the medulla oblongata and/or cervical cord. During surgery we found: 1) poor quality of omentum in most cases; 2) degenerative changes in the cervical spine; 3) anatomical anomalies at the V4 segments of the vertebral arteries; 4) moderate to severe atherosclerosis at both V4 segments; 5) unilateral absence or stenosis in the anterior-ventral spinal arteries (AVSAs). All patients received omentum on the anterior, lateral and posterior surface of the medulla oblongata, and in 9 cases, an additional segment at the C5-C6 level.
Neurological improvement was better during the first days or weeks after surgery than in the following months or years, in all patients. 13 patients suffered neurological impairment in about 4 months later, due to greater deterioration of the cervical spine, by contrast, 7 patients with mild ALS have experienced neurological improvement by 80 to 100% during a follow-up of 4 and 6 years.
These results confirm that ALS is of ischemic origin in the intraparenchymal territory of the AVSAs and/or in anterior spinal artery caused by atherosclerosis and associated to anatomical variants in the V4 segments of the vertebral arteries. Because in contrast to this, its revascularization by means of omentum can cure (mild degree) or improve this disease” [26].
Kamalesh Dase et al., “The study was designed to evaluate the role of environmental, occupational, and familial risk factors in development of ALS.
This was a case control study of 110 cases of definite ALS with 240 age and sex matched controls. Investigations were done on the following aspects- family history, occupation, living place, source of drinking water, exposure to industrial, chemical, agricultural toxins and heavy metals, physical and electrical injury, working under magnetic field for more than 10 years in both the groups. Clinical examinations, electrophysiological, and neuroimaging studies were done in every patient. Chi square test, logistic regression analysis, and calculation of odds ratio were used to analyze the data.
Rural livings (odds ratio = 1.99), smoking (odds ratio = 1.88), insecticides, and pesticides exposures (odds ratio = 1.61), electrical injury (odds ratio = 6.2) were detected as the associated factors in development amyotrophic lateral sclerosis.History of electric shock or injury was present in eight (7.3%) cases and in three (1.3%) controls. A strong association has been detected between the electric shock or injury and development of ALS (P = 0.003, OR = 6.20, 95% CI = 1.75-21.98)’’ [27].
Malek AM et al., “Exposure to pesticides and agricultural chemicals has been linked to amyotrophic lateral sclerosis (ALS) although findings have been inconsistent. A meta-analysis of studies published through May, 2011 was conducted to investigate the association of pesticide exposure and risk of ALS.
Six peer-reviewed studies that met criteria were included in a meta-analysis of men involving 1,517 ALS deaths from one retrospective cohort study and 589 ALS or motor neuron disease cases from five case-control studies. A random effects model was used to calculate sex-specific pooled odds ratios (ORs).
Evidence was found for an association of exposure to pesticides and risk of ALS in male cases compared to controls (OR=1.88, 95% CI: 1.36-2.61), although the chemical or class of pesticide was not specified by the majority of studies.
This meta-analysis supports the relationship of exposure to pesticides and development of ALS among male cases compared to controls. The weight of evidence links pesticide exposure to ALS; additional prospective studies with a target exposure group are necessary to better elucidate the relationship. Future research should focus on more accurate exposure assessment and the use of job exposure matrices” [28].
Zhou H et al., “To estimate the relationship between exposure to extremely low-frequency electromagnetic fields (ELF-EMF) and the risk of amyotrophic lateral sclerosis (ALS) by a meta-analysis.
Through searching PubMed databases (or manual searching) up to April 2012 using the following keywords: “occupational exposure”, “electromagnetic fields” and “amyotrophic lateral sclerosis” or “motor neuron disease”, seventeen studies were identified as eligible for this meta-analysis. The associations between ELF-EMF exposure and the ALS risk were estimated based on study design (case-control or cohort study), and ELF-EMF exposure level assessment (job title or job-exposure matrix). The heterogeneity across the studies was tested, as was publication bias.
Occupational exposure to ELF-EMF was significantly associated with increased risk of ALS in pooled studies (RR = 1.29, 95%CI = 1.02-1.62), and case-control studies (OR = 1.39, 95%CI = 1.05-1.84), but not cohort studies (RR = 1.16, 95% CI = 0.80-1.69). In sub-analyses, similar significant associations were found when the exposure level was defined by the job title, but not the job-exposure matrix. Significant associations between occupational exposure to ELF-EMF and increased risk of ALS were found in studies of subjects who were clinically diagnosed but not those based on the death certificate. Moderate heterogeneity was observed in all analyses.
Our data suggest a slight but significant ALS risk increase among those with job titles related to relatively high levels of ELF-EMF exposure. Since the magnitude of estimated RR was relatively small, we cannot deny the possibility of potential biases at work. Electrical shocks or other unidentified variables associated with electrical occupations, rather than magnetic-field exposure, may be responsible for the observed associations with ALS” [29].
Banack SA, “Beta-methylamino-L-alanine (BMAA) occurs in higher levels in museum specimens of the Guamanian flying fox than in the cycad seeds the flying foxes feed on, confirming the hypothesis that cycad neurotoxins are biomagnified within the Guam ecosystem. Consumption of a single flying fox may have resulted in an equivalent BMAA dose obtained from eating 174 to 1,014 kg of processed cycad flour. Traditional feasting on flying foxes may be related to the prevalence of neuropathologic disease in Guam” [30].
Cox PA et al., “We here report biomagnification (the increasing accumulation of bioactive, often deleterious molecules through higher trophic levels of a food chain) of the neurotoxic nonprotein amino acid beta-methylamino-l-alanine (BMAA) in the Guam ecosystem. Free-living cyanobacteria produce 0.3 microg/g BMAA, but produce 2-37 microg/g as symbionts in the coralloid roots of cycad trees. BMAA is concentrated in the developing reproductive tissues of the cycad Cycas micronesica, averaging 9 microg/g in the fleshy seed sarcotesta and a mean of 1,161 microg/g BMAA in the outermost seed layer. Flying foxes (Pteropus mariannus), which forage on the seeds, accumulate a mean of 3,556 microg/g BMAA. Flying foxes are a prized food item of the indigenous Chamorro people who boil them in coconut cream and eat them whole. Chamorros who die of amyotrophic lateral sclerosis/parkinsonism-dementia complex (AL-SPDC), a neurodegenerative disease with symptoms similar to amyotrophic lateral sclerosis, Parkinson’s disease, and Alzheimer’s disease, have an average of 6.6 microg/g BMAA in their brain tissues. The biomagnification of BMAA through the Guam ecosystem fits a classic triangle of increasing concentrations of toxic compounds up the food chain. This may explain why the incidence of ALS-PDC among the Chamorro was 50-100 times the incidence of amyotrophic lateral sclerosis elsewhere. Biomagnification of cyanobacterial BMAA may not be unique to Guam; our discovery of BMAA in the brain tissue from Alzheimer’s patients from Canada suggests alternative ecological pathways for the bioaccumulation of BMAA in aquatic or terrestrial ecosystems” [31].
Caroline Ingre et al., “(ALS) is the most common motor neuron disease. It is typically fatal within 2–5 years of symptom onset. The incidence of ALS is largely uniform across most parts of the world, but an increasing ALS incidence during the last decades has been suggested. Although recent genetic studies have substantially improved our understanding of the causes of ALS, especially familial ALS, an important role of non-genetic factors in ALS is recognized and needs further study. In this review, we briefly discuss several major genetic contributors to ALS identified to date, followed by a more focused discussion on the most commonly examined non-genetic risk factors for ALS. We first review factors related to lifestyle choices, including smoking, intake of antioxidants, physical fitness, body mass index, and physical exercise, followed by factors related to occupational and environmental exposures, including electromagnetic fields, metals, pesticides, β-methylamino-L-alanine, and viral infection. Potential links between ALS and other medical conditions, including head trauma, metabolic diseases, cancer, and inflammatory diseases, are also discussed. Finally, we outline several future directions aiming to more efficiently examine the role of non-genetic risk factors in ALS.
Occupations / environmental factors
Workers in various occupations with seemingly disparate exposures have been reported to be potentially at altered risk of ALS, including athletes, carpenters, cockpit workers, construction workers, electrical workers, farm workers, hairdressers, house painters, laboratory technicians, leather workers, machine assemblers, medical service workers, military workers, nurses, power production plant workers, precision metal workers, programmers, rubber workers, shepherds, tobacco workers, veterinarians, and welders. These occupations potentially involve work exposures to chemicals, pesticides, metals, and electromagnetic fields (EMF). Common denominators among these different occupations are not easily identified.
Military personnel are exposed to a battery of unique and potentially harmful factors, including physical and psychological exertion and trauma, transmissible agents (eg, viruses) and vaccines, toxic substances (eg, heavy metals and chemicals), and other environmental toxicants specific to particular deployment Qareas. A review article focusing on the potential links between military-related factors and ALS has been published recently, and concluded that although there is evidence suggesting a role of military service in ALS, it is too premature to draw a firm conclusion regarding a causal relationship.
Electric occupation, electric shock, and electromagnetic field
ALS has been associated with “electrical” occupations, especially welding. Magnetic fields, electrical fields, contact currents, microshocks, and both perceptible and imperceptible electric shocks all contribute to occupational exposure to extremely low frequency EMF. The reported association of ALS with EMF is generally weaker than that with electrical occupations. Evidence is not yet available to distinguish whether electric shocks or exposure to EMF underlies the association between electrical occupation and ALS. A meta-analysis suggested that there might be a slight but statistically significant increase in ALS risk among people with job descriptions related to relatively high levels of EMF exposure. Studies using residential proximity to power lines as a proxy for EMF exposure have failed to support such a relationship. Different exposure levels investigated in studies of occupational, compared with residential, exposure to EMF may partly explain the different findings to date.
Metals
That lead may be a culprit in ALS etiology is a long-standing hypothesis. Previous studies have mostly supported this relationship, relying in general on indirect measures of lead exposure. Lead levels in both blood and bone were found to be associated with ALS, although others found only an association for blood and not bone. Blood lead levels may reflect current environmental lead exposure and may also reflect mobilization of lead from bone. Lead toxicokinetics and bone metabolism may therefore modify the lead-ALS association. A recent case-control study observed that blood lead levels were high among ALS cases compared with controls, even after careful adjustment for bone turnover status and a polymorphism affecting lead toxicokinetics.
The neurotoxic properties of manganese are well known. Manganese crosses barrier systems at the choroid plexus and accumulates in the central nervous system, with a longer half-life in nervous tissue. Welders exposed to manganese demonstrate motor impairments in general and impaired fine motor skills specifically. Manganese concentrations in CSF samples of ALS patients were significantly elevated (median 5.67 μg/L) compared with healthy controls (median 2.08 μg/L). Furthermore, manganese concentrations were higher in the CSF than in the plasma of ALS patients, suggesting a transport of manganese into the central nervous system in ALS.
Iron serves as a cofactor for regulatory enzymes in the electron transport chain in the mitochondria. Brain iron content increases with age, and iron accumulation has been noted in other neurodegenerative disorders. Among ALS patients, increased iron concentration has been reported in the ventral spinal cord and in the motor cortex, especially the hand knob region, presumably corresponding to the small hand muscle weakness seen in these patients.
The potential role of the metalloid selenium has been investigated in endemic clusters of ALS in selenium-rich regions of South Dakota and in Italy. Recent studies of selenium in the CSF of patients in Italy have shown elevated selenite concentrations, maybe related to elevated selenium exposure via drinking water. To what extent these findings may be generalizable to other population remains to be elucidated.
Other metals with potential relevance for ALS are copper, aluminum, arsenic, cadmium, cobalt, zinc, vanadium, and uranium, all of which have been found in significantly elevated concentrations in the CSF of ALS patients when compared with healthy controls.
Pesticides are in widespread use worldwide and can be measured in various concentrations in air, food, and water. An association between pesticide use and ALS has been explicitly evaluated and suggested in previous studies, including two recent studies from India and the USA (organochlorine compounds, pyrethroids, herbicides, and fumigants specifically). In meta-analyses, pesticide use was found to be significantly associated with a higher risk of ALS, although the latter study169 suggested a male-specific association.
Exposure to β-methylamino-L-alanine (BMAA), an atypical amino acid, has been proposed to explain the high incidence of ALS-PDC in the western Pacific. BMAA was originally believed to be produced on Guam and elsewhere by a local Micronesian plant, Cycas micronesica; more recently, it was recognized that BMAA does not originate from the plant itself but rather from cyanobacteria.
Further, cyanobacteria apparently produce BMAA in settings outside Guam. In one study, BMAA was higher in the brain and spinal cord tissues of patients with ALS or Alzheimer’s disease than in healthy controls or patients with other diseases. A recent study has demonstrated that BMAA bioaccumulates in the Baltic Sea ecosystem and identified pathways for human exposure. Neurotoxins other than BMAA produced by cyanobacteria may also contribute to neurodegeneration.
Previous viral infection has also been considered as a potential risk factor for ALS. For example, a role of enteroviral infections in ALS has been hypothesized since neurons in the anterior horn of the spinal cord are the target cells both in ALS and enteroviral infections, including poliomyelitis. Using reverse transcriptase (RT) in situ polymerase chain reaction, enterovirus RNA was detected in motor neurons of the anterior horn of patients with ALS. Exposure to other viruses may also be important. Human herpesvirus (HHV)-6 seropositivity was associated with a more than threefold risk of ALS, and HHV-8 seropositivity with a more than eightfold risk. Retroviruses, such as human immunodeficiency virus and human T-cell lymphotrophic virus-1, caused motor neuron syndromes. Some as yet unidentified retrovirus might also be a risk factor for ALS, because a mouse retrovirus (murine leukemia virus) causes both a lower motor neuron syndrome and leukemia/lymphoma. A more broad measure of retroviral infection, ie, serum activity of reverse transcriptase (an enzyme characterizing retroviral infections), was similar among ALS patients and their blood relatives, but lower among their spouses, who had levels similar to that of other non-blood-related controls. More recent studies of the expression of human endogenous retroviral sequences have revealed significantly increased expression of human endogenous retro virus type K (HERV-K), one of the two most studied human endogenous retroviruses given its complete open reading frame and ability to form virus-like particles, in the serum, muscle, and post mortem brain tissue of ALS patients.
The general belief that ALS is a complex multifactorial disease has suggested the importance of studying the relationship between ALS and other medical conditions, which may share environmental risk factors or a genetic predisposition with ALS. In this review, we focus our discussion on the potential roles of head trauma, metabolic diseases, cancer, and neuroinflammation in ALS.
Early case-control studies reported a significant association between history of head trauma and ALS. Aiming to rule out the possibilities of recall bias and reverse causality (ie, trauma as a result, rather than a cause, of ALS), later studies generally used more objective assessment of head trauma history and excluded traumas experienced during the years immediately before the diagnosis of ALS. Severe head traumas that were hospitalized were not associated with a higher risk of ALS in Sweden. A possible association of ALS with milder head traumas, perhaps specifically with repeatedly experienced mild traumas, has not been thoroughly addressed.
An interest in the relationship between metabolic disorders and ALS arose after the observation that ALS patients are hypermetabolic. Previous studies suggested that type 2 diabetes is associated with a lower risk of ALS, while type 1 diabetes, as well as some other autoimmune diseases, might instead be risk factors for ALS. In a recent study, we confirmed an inverse association between type 2 diabetes and risk of ALS, and found that type 1 diabetes was indeed associated with a threefold risk of ALS. Medications used for treatments of metabolic disorders, independently of the underlying disorders, may also be associated with the development of ALS, the evidence gathered to date is inconclusive regarding the relationship between use of statins and the risk or progression of ALS, and between the antidiabetic drug, pioglitazone, and progression of ALS. These findings are not surprising given the complexity of their properties. Pioglitazone, for example, is both antioxidant and anti-inflammatory and may protect against neurodegeneration, but it is also antidiabetic and antidyslipidemic and may therefore be detrimental for ALS, if the emerging evidence of a potentially protective effect of obesity and type 2 diabetes with regard to ALS proves true.
Although the incidence of neurodegenerative diseases and of cancer increases in older adults, these two groups of diseases are characterized by largely opposing cellular behavior, ie, premature cell death in neurodegeneration and resistance to cell death in carcinogenesis. A potential inverse relationship between neurodegenerative diseases (eg, Alzheimer’s disease and Parkinson’s disease) and cancer has been observed, and is plausible for the reasons discussed. Based on clinical case series or case reports, earlier studies suggested a positive association between ALS and cancer. Most epidemiological studies have refuted such a link in general, except for melanoma. The most recent, large-scale, prospective cohort studies have similarly refuted a positive association between cancer, including melanoma, and ALS.
Neuroinflammation
Since the earliest pathological changes in ALS appear to occur in axons, dendrites, and synapses,206 studies of the relationship between inflammatory conditions around the motor unit and ALS may shed light on the pathological development of ALS. Clinically, early symptoms of ALS can be difficult to differentiate from symptoms of other inflammatory neuromuscular diseases such as myositis, myasthenia gravis, Guillain–Barré syndrome, and multiple sclerosis. Due to the difficulties in determining the correct diagnosis, misdiagnosis may be an explanation for any higher-than-expected co-occurrence of ALS and inflammatory diseases. Interestingly, ALS and multiple sclerosis were reported to co-occur in individuals with the C9ORF72 repeat expansion, suggesting some biological overlaps between ALS and autoimmune/inflammatory diseases. Apart from several reports of cases diagnosed with both ALS and with some of the conditions above, few studies have addressed this issue” [32].
Han-Xiang Deng et al., “(ALS) is a fatal disorder of motor neuron degeneration. Most cases of ALS are sporadic (SALS), but about 5-10% of ALS cases are familial (FALS). Recent studies have shown that mutations in FUS are causal in approximately 4-5% of FALS and some apparent SALS cases. The pathogenic mechanism of the mutant FUS-mediated ALS and potential roles of FUS in non-FUS ALS remain to be investigated.
Immunostaining was performed on postmortem spinal cords from 78 ALS cases, including SALS (n=52), ALS with dementia (ALS/dementia, n=10) and FALS (n=16). In addition, postmortem brains or spinal cords from 22 cases with or without frontotemporal lobar degeneration (FTLD) were also studied. In total, 100 cases were studied.
FUS-immunoreactive inclusions were observed in spinal anterior horn neurons in all sporadic and familial ALS cases, except for those with SOD1 mutations. The FUS-containing inclusions were also immunoreactive with antibodies to TDP43, p62 and ubiquitin. A fraction of tested FUS antibodies recognized FUS inclusions and an unusual antigen retrieval appeared to be important for detection of the skein-like FUS inclusions.
Although mutations in FUS account for only a small fraction of FALS and SALS, our data suggest that FUS protein may be a common component of the cellular inclusions in non-SOD1 ALS and some other neurodegenerative conditions, implying a shared pathogenic pathway underlying SALS, non-SOD1 FALS, ALS/dementia and related disorders. Our data also indicate that SOD1-linked ALS may have a distinct pathogenic pathway from SALS and other types of FALS.
(ALS) is a fatal paralytic disorder caused by degeneration of motor neurons in the brain and spinal cord. Most cases of ALS are of unknown etiology and appear as sporadic ALS (SALS). About 5-10% of ALS cases have a family history. Some forms of familial ALS (FALS) are linked to genetic mutations in specific genes. Mutations in the Cu/Zn superoxide dismutase gene (SOD1) account for approximately 20% of FALS cases 1, 2. Mutations in the TAR DNA-binding protein gene (TARDBP, or TDP43) may occur in about 3-4% of FALS 3-5. Rare genetic defects in several other genes have been found in ALS or ALS-like motor neuron diseases 6. Recently, mutations in FUS have been identified in 4-5% of FALS cases 7, 8 and some SALS cases.Altogether, mutations in specific genes have been identified in about 30% of the FALS cases. The causes for the remaining FALS cases have yet to be identified.
In contrast to FALS, the etiology and the pathogenic mechanisms underlying SALS, the most common form of ALS, remain largely unknown. Understanding the causes and pathogenic mechanisms of SALS remains to be a major challenge.
Protein aggregation has been recognized as a pathological hallmark in several neurodegenerative disorders, such as extracellular amyloid-beta plaques and intracellular tau neurofibrillary tangles in Alzheimer disease, and alpha-synuclein containing Lewy bodies in Parkinson disease 15. In ALS, protein aggregates are most common in spinal motor neurons and the ubiquitin-immunoreactive and skein-like aggregates are thought to be pathognomic of ALS. SOD1-immunoreactive aggregates are a prominent pathology in SOD1-mediated ALS, but not in SALS 18. TDP43-immunoreactive aggregates have been identified in SALS, but not in SOD1-ALS. Both TDP43 and FUS are DNA/RNA-binding proteins. They are predominantly present in cell nuclei in physiological condition. In neurons with cytoplasmic TDP43 aggregates, nuclear TDP43 is diminished. It is currently unknown if FUS is involved in the pathogenesis of SALS and other forms of FALS without FUS mutations.
In this study, we analyzed postmortem tissues using immunostaining to explore post-translational involvement of FUS in different types of ALS and other neurodegenerative disorders, as well as neurological disease-free controls’’[33].
Ross CA ET AL., “Neurodegenerative diseases such as AD, PD, Huntington’s disease (HD), (ALS) and prion diseases are increasingly being realized to have common cellular and molecular mechanisms including protein aggregation and inclusion body formation.
The aggregates usually consist of fibers containing misfolded protein with a beta-sheet conformation, termed amyloid. There is partial but not perfect overlap among the cells in which abnormal proteins are deposited and the cells that degenerate. The most likely explanation is that inclusions and other visible protein aggregates represent an end stage of a molecular cascade of several steps, and that earlier steps in the cascade may be more directly tied to pathogenesis than the inclusions themselves. For several diseases, genetic variants assist in explaining the pathogenesis of the more common sporadic forms and developing mouse and other models. There is now increased understanding of the pathways involved in protein aggregation, and some recent clues have emerged as to the molecular mechanisms of cellular toxicity. These are leading to approaches toward rational therapeutics” [34].
Huss A et al., “(ALS) has been associated with exposures in so-called ‘electrical occupations’. It is unclear if this possible link may be explained by exposure to extremely low-frequency magnetic fields (ELF-MF) or by electrical shocks. We evaluated ALS mortality in 2000-2008 and exposure to ELF-MF and electrical shocks in the Swiss National Cohort, using job exposure matrices for occupations at censuses 1990 and 2000. We compared 2.2 million workers with high or medium vs. low exposure to ELF-MF and electrical shocks using Cox proportional hazard models. Results showed that mortality from ALS was higher in people who had medium or high ELF-MF exposure in both censuses (HR 1.55 (95% CI 1.11-2.15)), but closer to unity for electrical shocks (HR 1.17 (95% CI 0.83-1.65)). When both exposures were included in the same model, the HR for ELF-MF changed little (HR 1.56), but the HR for electric shocks was attenuated to 0.97. In conclusion, there was an association between exposure to ELF-MF and mortality from ALS among workers with a higher likelihood of long-term exposure” [35].
Sarah Martin et al., “ALS is a neurodegenerative disease predominantly affecting upper and lower motor neurons, resulting in progressive paralysis and death from respiratory failure within 2 to 3 years. The peak age of onset is 55 to 70 years, with a male predominance. The causes of ALS are only partly known, but they include some environmental risk factors as well as several genes that have been identified as harbouring disease-associated variation. Here we review the nature, epidemiology, genetic associations, and environmental exposures associated with ALS.
Genetics and ALS, There are now more than 25 genes in which an association with ALS has been replicated, with the rate of gene discovery doubling every 4 years (http://alsod.iop.kcl.ac.uk) 69. In up to 10% of people, there is a family history of ALS in a first-degree relative, but detailed genealogical studies extending to more distant relatives and including related diagnoses suggest that more than 20% have a relevant family history. The genes responsible for familial ALS have now been identified for about 70% of all cases, but there is a significant genetic component, even in those without a family history. Twin studies suggest the heritability is about 60%, and nearly every familial ALS gene has also been implicated in apparently sporadic ALS 70, 71. Furthermore, statistical analysis shows that the distinction between familial and sporadic ALS is not clear-cut, and large-scale genome-wide association studies (GWAS) show that the genetic architecture of sporadic ALS is one in which rare variation, more usually associated with familial disease, is disproportionately important.
The most recent GWAS of ALS identified four new associations, 3 of which were successfully replicated. An interesting feature of the study was that even though this was a study of people with apparently sporadic ALS, there were associations in genes previously identified from family-based studies – C9orf72, TBK1, and NEK1 – further supporting the notion that familial and sporadic ALS are not mutually exclusive categories but rather a spectrum. These 3 genes all harbour variants that are moderately penetrant. In other words, carrying a disease-associated variant does not mean ALS will inevitably follow. Current thinking is that common diseases are the consequence of the additive effects of small increases in risk from multiple common variations (polygenic), and rare diseases are the consequence of single gene variants that are themselves rare but have a large effect on the probability of disease (monogenic). For example, height and schizophrenia are polygenic traits, while Huntington’s disease and Kennedy’s disease are monogenic diseases. ALS sits somewhere between these two extremes, with a lifetime prevalence that is far greater than is typical for a monogenic disease but far less than a common disease, and it is perhaps, therefore, to be expected that its genetic architecture also seems to sit somewhere between polygenic effects and monogenic high-penetrance disease.
There are 3 genes that have had a major impact on our understanding of ALS. ALS-linked dominant mutations in the superoxide dismutase gene SOD1 were first identified in 1993, and since then mutations have been found in every exon of the gene. The SOD1 protein is a free radical scavenger, and loss of function, increasing free radical damage in cells, is a logical hypothesis to consider. Several well-characterised SOD1 variants do not lead to a reduction in dismutase activity, and the evidence instead supports a toxic gain of function. Transgenic SOD1 mice develop a motor neuron degeneration and have been used to model the disease for treatment development 79. The second important ALS gene is TARDBP, which codes for TDP-43, a protein regulating RNA expression and the major component of intracellular inclusions in ALS. The discovery of ALS-linked mutations in this gene was the first of many showing RNA processing defects to be important in ALS pathogenesis and, importantly, showed that the TDP-43 inclusions were not simply a passive marker of neuronal death but a crucial part of the disease pathway. The third important genetic finding in ALS was of linkage and then association of a locus on chromosome 9, which led researchers to the identification of a massive expansion of a hexanucleotide repeat in intron 1 of the C9orf72 gene. This is the most frequent cause of ALS, being responsible for about 30% of familial and up to 10% of sporadic cases.
The focus of genetic research in ALS in the immediate future is therefore on rare variation. This is best discovered through high-throughput sequencing, and this technique has already identified several familial ALS genes. The major challenge facing researchers is how to interpret the findings, since the identification of a rare variant in an ALS gene is not in itself strong evidence of relevance in that individual, and over-representation of rare variation in cases over controls in a particular gene does not provide sufficient information for genetic counselling on a specific variant. The amount of heritability explained by genetic information captured on genome-wide microarrays is about 12%, implying that the remainder is in rare variants and other types of genetic variation such as copy number variation, microsatellite repeats, post-transcriptional RNA editing, and epigenetic changes. These are likely to be the next targets of ALS genetics research and are reliant on international research consortia. Project MinE is one such global collaboration that aims to analyse DNA from at least 15,000 people with ALS and 7,500 controls.
Evidence of an immuno-inflammatory component in ALS pathogenesis is compelling. A pathological hallmark of the neuroinflammation is prominent microglia activation at involved sites. T-regulatory lymphocytes (Tregs) are important immunomodulatory cells that regulate the balance between activation and suppression of the immune response and control the microglia in the central nervous system: specifically, pushing them towards a state in which remodeling and repair activities are activated. Defects in Treg levels or function have been found in ALS patients, becoming more frequent as the disease progresses. Treg levels are inversely correlated with disease severity, so that lower levels are seen in more severe disease, and survival is worse in those with Treg defects. Studies are now underway to explore immune therapies that might improve Treg function and therefore improve survival.
Poliovirus and other enteroviruses can cause a post-infectious myelitis with subsequent paralysis, and HIV infection can result in an ALS-like syndrome. Studies of serum and cerebrospinal fluid from ALS patients suggested that an activated endogenous retrovirus was associated with ALS. Recently, the sequence has been identified as possibly HERV-K, an endogenous retrovirus that exists as an open reading frame in the human genome. In mice, the env protein component of HERV-K is toxic to motor neurons. There is no evidence that HERV-K is causative of the disease in humans, but studies are now underway to explore if antiretrovirals might slow progression and improve survival in ALS.
In contrast to genetics, environmental risk factors for ALS have been more difficult to identify. Such studies are expensive to perform but difficult to fund and are heavily reliant on recall. As a result, they are susceptible to bias. Unlike genome-wide analyses, in which a hypothesis-generating approach can be taken, it is not straightforward to assay all possible environmental factors, and so a selected subset of assumed risk factors is tested. Smoking has been associated with increased risk of ALS in some studies and may hold a higher risk in some subgroups. Occupation, particularly military service with deployment, has been associated with risk of ALS, but the evidence mainly comes from the US, where there are large military datasets. Physical activity is another widely studied risk factor, partly because of a number of high-profile sports players who have had ALS and because of people with ALS having a low BMI on presentation and higher levels of leisure sports participation. It is not clear whether having higher levels of physical activity raises the risk of ALS and, if it does, whether it is the activity itself or being genetically predisposed to high sporting prowess that is the mechanism. Similarly, electric shock is not a risk factor in some analyses but is in others. There is mixed evidence for the involvement of chemicals, such as heavy metals, ambient aromatic hydrocarbons, pesticides, and cyanotoxins. Trauma, including head injury, also appears to be a risk factor in meta-analysis’’ [36].
Kumar Abhinav et al., “The evidence from this systematic review clearly indicates that there is a syndrome of non‐progressive spinal cord damage after electrical injury. This syndrome is often associated with site of onset at the entry or exit point of the current, and is associated with more severe electrical injury. Patients with non‐progressive syndromes may recover partially or completely. Severe electrical injury, including lightning strike, is predictive of a more rapid onset of symptoms and signs for all syndromes. Severe electrical injury predicted non‐progressive syndromes whereas cases with progressive syndromes including ALS reported in the literature had mild electrical injury. The most parsimonious explanation is that stronger shocks result in myelopathy, and the ALS cases associated with mild shock are then a result of recall and publication bias. Furthermore, age at shock predicted delay in disease onset, with a younger age at shock associated with a longer delay in disease onset. This effect of age at shock was also true for non‐progressive syndromes and was therefore not an effect of the later age at onset of ALS. The explanation of the relationship between younger age at shock and longer interval between shock and disease onset is uncertain and may be related to the altered vulnerability of the ageing nervous system. Further, for 35 of the 44 cases with ALS, for whom there was information, the median age at onset of ALS after the electrical injury was 36 years. This earlier age at onset of ALS is most probably related to occupational bias, as young men are more likely to be employed as electricians and experience electric shocks at work.
In most cases classified as ALS for which information was available, the site of onset of the disease was either at the entry or exit point, suggesting a causal relationship between injury and neurological disease. It is, difficult to explain the significance of this observation, as in 25 of the 44 cases in this group sufficient information was lacking, and this relationship was also true for non‐progressive syndromes. Thus no firm conclusions can be drawn about the relationship between site of injury and type of syndrome (ALS vs non‐progressive disease).
For all progressive motor neurone syndromes, the median interval between electrical injury and disease onset was 2.25 years. This contrasts with the non‐progressive syndromes in which the median delay between electrical injury and the onset of disease was only just over 1 week. This suggests that the patients classified with progressive syndromes are heterogeneous. In non‐progressive cases in which neurological deficits follow electrical injury within a few days or a week, a causal relationship is likely.
There is a strong male predominance (93% male, 7% female), which may reflect an occupational bias. Of greater interest is the paucity of patients with bulbar onset in the progressive syndrome group. For 33 patients with a progressive syndrome for whom information on site of onset was available, only 2 had bulbar onset. In most clinic‐based and population‐based studies, the proportion of bulbar‐onset ALS cases is about 25% 16. The rarity of bulbar onset in the progressive group may be related to the sex ratio in cases reviewed here, as elderly women are more likely to have bulbar‐onset ALS, 17 but this cannot be the complete explanation. This apparent under‐representation of bulbar‐onset disease was also evident in the non‐progressive syndromes, most likely because the neurological syndrome is related to the site of injury, which is usually in the limbs.
Unfortunately, there is relatively little information on neuropathological changes associated with electric shock as related to the syndromes described in this study. Furthermore, among all the cases we have reviewed, in only three was ALS pathologically proved (<3%). A variety of mechanisms, including excitotoxicity and microglial activation, might contribute to ongoing damage if electrical injury were acting as a trigger factor for ALS. Electrical injury may cause a wide range of morphological changes in the central nervous system. Cellular changes include neuronal chromatolysis, neuronophagia and neuronal loss. Microglial activation, which is an early event in central nervous system damage, is prominent, as is infiltration of blood‐borne macrophages and neutrophils. Electrical injury might thus be expected to trigger a cascade of cellular damage in individuals at risk of developing ALS—for example, those carrying SOD1 gene mutations. Even sporadic ALS may be associated with genetic risk factors, and the increased risk of ALS in military personnel or other occupational groups might be related to genetic selection. Thus, the notion that injury might trigger a cascade of neuronal damage is applicable to both familial and sporadic ALS. The fact that our review does not support this concept is therefore somewhat surprising, but may suggest that such cascades are intrinsically generated and propagated and are not significantly influenced even by major focal insults.
2 population‐based case–control studies found no association between electric shock and ALS; in particular, in the study conducted in Western Washington State no statistically significant association was found and this quality of finding was deemed to be high in a subsequent review Similarly, the Scottish Motor Neurone Disease Register Case–Control Study demonstrated no significant difference between patients and controls in number of electric shocks; the quality of these findings were rated lower owing to lack of actual numbers and confidence intervals. These studies highlight the difficulties in designing a case–control study to provide high‐quality evidence in support of risk factors affecting a small number of patients because, apart from an increased likelihood of bias, small patient numbers do not lead to generation of reliable dose–response data.
Unquestionably, the most comprehensive review of the neurological sequelae of electrical trauma to date is that of Panse, 24 but this does not provide a systematic analysis of the relationship between electrical injury and ALS. Panse noted that ALS is extremely rare in victims of electrical accidents and quotes Pietrusky (quoted by Panse) who studied the records of 797 victims of electric accidents and found only one relevant case. Panse quotes that “certainly, this harvest of spinal atrophic sequelae with a progressive course and (usually) spastic components culled from the whole world literature is not very impressive”; he, believed that he could not exclude a causal relationship between electric trauma and a syndrome identical with ALS.
Our systematic review of the literature on the relationship between ALS and electrical injury indicates that a syndrome of non‐progressive spinal cord damage, often with both LMN and UMN components, is strongly linked to more severe shock, and that the onset of disease is closely related to time from the injury. A well‐defined entry or exit point is common in such cases.
The relationship between electric injury and ALS is less certain, and the evidence reviewed does not support a causal relationship between ALS and electric shock. Only systematic prospective studies could resolve this, but epidemiological evidence, although incomplete, does not suggest that electrical trauma is associated with increased risk of developing ALS. ALS is a heterogeneous disorder and it is possible that in some individuals electrical trauma (or other forms of trauma) could trigger the disease process’’ [37].
Aurélie Delzor et al., “Historically, an association has been observed between a mutation on the superoxide dismutase 1 gene (SOD1) and FALS. 2 other mutations 3–8 have since been discovered, as C9 or f72 (chromosome 9 open reading frame 72), TARDBP (TDP-43 encoding gene) and FUS (fused in sarcoma protein) are commonly identified in FALS cases. Although SOD1, FUS and TARDBP mutations have also been found in SALS cases,2 17 the current broad scientific consensus is in favour of a gene–environment interaction causing SALS: lifestyle factors, environmental exposure, occupational exposure and handling toxic compounds are among the many factors that can play a role in the appearance of the pathology. Among lifestyle factors, smoking is the factor that has been most documented and is mainly associated with a higher risk of ALS, whereas coffee and alcohol consumption are considered protective or not associated with ALS. Other associations have been proposed as occupational exposure to electromagnetic fields, frequent head trauma, contact with certain chemicals such as pesticides, formaldehyde, organic solvents and heavy metals.Another controversial hypothesis, often cited, is that physical activity, whether occupational or leisure related, is a risk factor for SALS. This theory is sustained by the higher risk of ALS in professional soccer players. On the Pacific island of Guam, ALS-Parkinsonism dementia complex (ALS-PDC), which presents similarly to ALS, occurred at 50 to 100 times the incidence seen worldwide in the 1950s. An epidemiological study established that consumption of a Chamorro diet was the only variable significantly associated with disease incidence. In 1967, Vega and Bell53 discovered a neurotoxin, β-N-methylamino-L-alanine (L-BMAA), in the genus Cycas, the seeds of which are used to make flour. Hence, L-BMAA could have been consumed by Chamorro people through multiple dietary sources, including cycad flour as well as meat from flying foxes and other animals that feed on cycad seeds. In the 1990s, L-BMAA was proposed as a cause of ALS-PDC.
This hypothesis is supported by the presence of L-BMAA in brain tissues of patients with ALS-PDC and ALS from Guam and Canada, as well as by its absence in controls.
In vitro and in vivo experiments also suggest that L-BMAA plays a role in neuropathological processes implicated in ALS. Indeed, the treatment of dissociated mixed spinal cord cultures with a concentration of L-BMAA around 30 μM caused selective motor neurone loss. Monkeys fed with large doses of the toxic acid from cycads developed neurological impairments:
Damaged motor neurones in the spinal cord produced a flaccid paralysis and then damaged neurones in the striatum and cortex, which produced Parkinsonism and behavioural changes. In rats, although the intraperitoneal injection of L-BMAA did not provoke any obvious motor dysfunction, it induced markers of oxidative stress in the liver and cellular changes in favour of apoptosis in motor neurones of the spinal cord In neonatal rats, L-BMAA induced significant systemic changes in energy metabolism and amino acid metabolism (identification of initial metabolite changes for lactate, acetate, D-glucose, creatinine and 3-hydroxybutyrate). Together, these findings suggest that acute toxicity of L-BMAA induces developmental alterations that result in long-term effects on brain function.
L-BMAA is also found to be associated with proteins in cyanobacteria and in brain tissue of patients with ALS. It has recently been proposed that L-BMAA may be misincorporated into proteins and thus may lead to protein aggregation, a hallmark of neurodegenerative diseases, inducing a chronic exposure to low levels of L-BMAA.
L-BMAA was found to be produced by a wide range of cyanobacteria; recently, it was shown that diatoms, the most common group of algae, could also produce it. The level of free or bound L-BMAA detected in cyanobacteria is controversial and the high concentrations reported in the first studies were challenged by several recent studies.
L-BMAA could be transferred from cyanobacteria or diatoms via zooplankton to organisms at higher trophic levels. Cox and collaborators have interestingly highlighted the biomagnification (increasing accumulation of bioactive, often deleterious, molecules through successively higher trophic levels of a food chain) of L-BMAA in trophic chain, explaining the large amounts detected in flying foxes from Guam. Owing to eutrophication and, to a lesser extent, climate changes, cyanobacterial blooms seem to be increasing in freshwater ecosystems worldwide. France is not exempt from this phenomenon as different genera of cyanobacteria are found on its territory.
Therefore, exposure of French patients with ALS to cyanobacteria, and thereby to cyanotoxins as L-BMAA, is a reasonable hypothesis that could potentially explain some ALS cases.
The French BMAALS program85 takes advantage of (1) the existing federation of BMAALS consortium members in the French network on ALS clusters detection and investigation, supported by INSERM (Institut National de la Santé et de la Recherche Médicale) and (2) of geoepidemiology to investigate patients’ environment (dwelling, occupational and leisure) in order to assess spatial association (not cause-and-effect) between ALS cases and a putative cyanobacterial exposure in combination with patients’ history about prior exposures.
A case–control study will be performed to investigate the potential routes of contamination by L-BMAA, which are: (1) ingestion of contaminated drinking water or dermal contact in recreational water; (2) consumption of aquatic or terrestrial food previously exposed to toxins; (3) cyanobacterial dietary supplements which are rich in protein content and (4) inhalation or aerosolisation.
To assess the exposure of patients to L-BMAA, 2 Delzor “ALS is the most common motor neurone disease. It occurs in two forms: familial cases, for which several genes have been identified and sporadic cases, for which various hypotheses have been formulated. Notably, the β-N-methylamino-L-alanine (L-BMAA) toxin has been postulated to be involved in the occurrence of sporadic ALS. The objective of the French BMAALS programme is to study the putative link between L-BMAA and ALS.
The programme covers the period from 1 January 2003 to 31 December 2011. Using multiple sources of ascertainment, all the incident ALS cases diagnosed during this period in the area under study (10 counties spread over 3 French regions) were collected. First, the standardized incidence ratio will be calculated for each municipality under concern. Then, by applying spatial clustering techniques, over incidence and under incidence zones of ALS will be sought. A case–control study, in the subpopulation living in the identified areas, will gather information about patients’ occupations, leisure activities and lifestyle habits in order to assess potential risk factors to which they are or have been exposed. Specimens of drinking water, food and biological material (brain tissue) will be examined to assess the presence of L-BMAA in the environment and tissues of ALS cases and controls.
It is of major importance to identify environmental risk factors causing SALS. The protocol presented here aims to study the link between L-BMAA and ALS in France by characterizing exposure modalities, either individual or collective, to cyanobacteria and more precisely to the L-BMAA toxin. Thanks to the questionnaire, it also intends to shed light on other assumptions formulated in literature as putative origins for SALS (as occupational exposure and sports practicing). Finally, our results could be used to generate a guide of precautions against behavioral risk leading to exposure to L-BMAA.
The results of this project should help to (1) give a clear picture of ALS distribution over 10 French counties; (2) identify clusters where environmental factors may play a greater role than elsewhere; (3) provide information about some environmental specificities of ALS clusters, especially regarding factors related to cyanobacteria presence and proliferation as also BMAA presence and (4) see to what extent the BMAA hypothesis seems to be relevant regarding the explanation of SALS clusters within the large French area considered. Despite limitations mainly due to i) interviews of patients’ relatives and ii) the controversy on L-BMAA analysis, this programme is of importance because it is the first to investigate the cyanobacteria hypothesis in France” [38].
Kevin A. Henry et al., “Few analyses in the United States have examined geographic variation and socioeconomic disparities in (ALS) incidence, because of lack of population-based incidence data. In this analysis, we used population-based ALS data to identify whether ALS incidence clusters geographically and to determine whether ALS risk varies by area-based socioeconomic status (SES). This study included 493 incident ALS cases diagnosed (via El Escorial criteria) in New Jersey between 2009 and 2011. Geographic variation and clustering of ALS incidence was assessed using a spatial scan statistic and Bayesian geoadditive models. Poisson regression was used to estimate the associations between ALS risk and SES based on census-tract median income while controlling for age, sex, and race. ALS incidence varied across and within counties, but there were no statistically significant geographic clusters. SES was associated with ALS incidence. After adjustment for age, sex, and race, the relative risk of ALS was significantly higher (relative risk (RR) = 1.37, 95% confidence interval (CI): 1.02, 1.82) in the highest income quartile than in the lowest. The relative risk of ALS was significantly lower among blacks (RR = 0.57, 95% CI: 0.39, 0.83) and Asians (RR = 0.63, 95% CI: 0.41, 0.97) than among whites. Our findings suggest that ALS incidence in New Jersey PLoS One. 2014; 9(5): e96556 [39] (Figure 16).
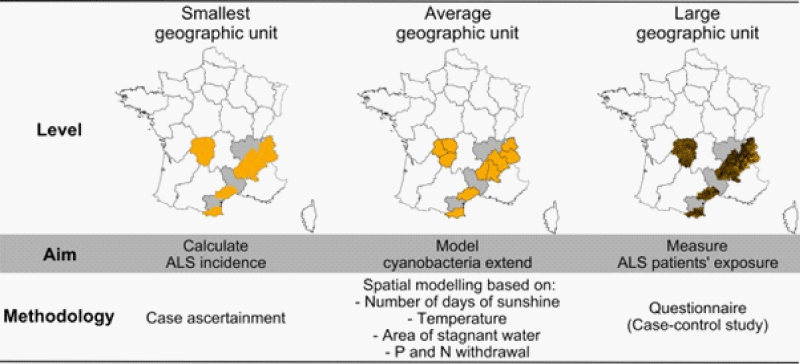
Figure 16: From reference: ALS incidence- cyanobacteria – ALS exposure.
James Rooney et al., “There has been much interest in spatial analysis of ALS to identify potential environmental or genetically caused clusters of disease. Results to date have been inconclusive. The Irish ALS register has been recently geocoded, presenting opportunity to perform a spatial analysis on national prospectively gathered data of incident cases over an 18-year period.
1,645 cases of ALS in Ireland from January 1995 to July 2013 were identified from the Irish ALS register. 1,638 cases were successfully geocoded. Census data from four censuses: 1996, 2002, 2006 & 2011 were used to calculate an average population for the period and standardized incidence rates (SIRs) were calculated for 3,355 areas (Electoral Divisions). Bayesian conditional auto-regression was applied to produce smoothed relative risks (RR). These were then mapped for all cases, males & females separately, and those under 55 vs over 55 at diagnosis. Bayesian and linear regression were used to examine the relationship between population density and RR.
Smoothed maps revealed no overall geographical pattern to ALS incidence in Ireland, although several areas of localized increased risk were identified. Stratified maps also suggested localized areas of increased RR, while dual analysis of the relationship between population density and RR of ALS yielded conflicting results, linear regression revealed a weak relationship’’ [40].
Logroscino G et al., “(ALS) epidemiology has rapidly developed in the last 30 years alongside the evolving changes in concepts in the field of clinical ALS and also due to the recent proposals of new classification system for motor neuron diseases. Many of these changes in the clinical scenario have been determined through the results of ALS population-based studies conducted in the last 20 years primarily in Europe. All the evidences converge to show that ALS risk is different across continents and ethnicities. In a European registry consortium named EURALS, ALS incident cases were drawn from a source population comprising almost 24 million people across Europe (ALS cases: 1028) and the estimated incidence was 2.2 per 100,000 person-years (py) for the general population. In contrast, other population-based studies have measured the lowest incidence in East Asia to be 0.89 per 100,000 py and in South Asia to be 0.79 per 100,000 py. A large part of Africa, Latin America and Asia does not have any population-based studies. The origin of geographic difference in ALS incidence is a matter of debate. Probably, this is partly due to genes (C9ORF72) and partly due to environmental risk factors. The rapid disappearance of ALS Foci in Guam, Kii, and West Guinea underline the importance of changes in lifestyle and environmental factors. The Global Burden of Disease, a project aiming to describe the burden of all diseases and injuries across all the countries of the world with a standardized protocol, has collected heterogeneous sources of data to estimate the burden of motor neuron diseases. The demographic changes related to increased expectation of life and the growth of the world population indicate that the load of motor neuron disease is rapidly moving toward 400 thousand prevalent cases. The burden is expected to shift toward Asia and Africa in the next decades for the rapid increase of expectation of life of countries with high demographic impact” [41].
Scott KM et al., “(ALS) is a degenerative disease of motor neurons that causes progressive paralysis and eventually results in death from respiratory failure. Environmental factors that trigger ALS might result in a pattern of geographical clustering of cases. We tested this hypothesis using the South-East England ALS population register, which covers south-east London, Kent and parts of neighbouring counties. The register’s catchment area was divided into postcode districts and sectors. The expected rates of ALS (adjusted for age and sex) were compared with the observed rates using a standardized residuals method and the SaTScan programme. Results: There were 406 cases of ALS identified in the catchment area during the study period. Four of the 126 postcode districts, all in Greater London, had residuals >2.5 SDs from the mean. Similarly, there were 15 postcode sectors (out of 420) that had residuals >1.96 SDs from the mean. Nine of these were in Greater London. SaTScan identified an area that had a 5.61-km radius in which the relative risk of ALS was 1.70 (p = 0.012). This area overlapped with the postcode districts and some of the sectors identified using the residuals method. These findings suggest an excess of ALS cases in some postcode districts in south-east England’’ [42].
Hatice Uenal et al., “Objective of this paper was to investigate the incidence, potential geographical clusters and the completeness of the (ALS) registry in Southern Germany (Swabia).
Age-standardized incidence rates (ASR) and ratios (SIR) as well as 95% confidence intervals (CI) were estimated at county level. Capture-recapture (CARE) procedures were applied taking data source dependency into account to estimate the quality of case ascertainment in the ALS registry Swabia. We identified 438 ALS cases (53% men, 47% women) in the target population of about 8.4 Mio inhabitants. The gender ratio (men∶ women) was 1.1∶1. The mean age at onset of ALS was 63.8 (SD = 11.9) years for men and 66.0 (12.2) for women. The age distribution peaked in the age group 70–74 years. The ASR of ALS was 2.5 per 100,000 person years (PY; 95% CI: 2.3–2.7). The mean SIR was 1.1 per 100,000 PY (95% CI: 1.0–1.2). High SIR suggesting geographical clusters were observed in two counties (Göppingen and Bodenseekreis), but the variation was not statistically significant (p-values = 0.2 and 0.5). The percentage of CARE estimated missing cases was 18.9% in the registry yielding an ASR of 3.1 per 100,000 PY. The high coverage of the CARE estimated completeness of the ALS registry Swabia indicates excellent quality for future projects. Regional variations have to be investigated further’’ [43].
Kokubo Y et al., “Between 1984 and 1996 we histopathologically examined 26 autopsy cases of (ALS) from the Mie Prefecture in eastern and southern Kii Peninsula, which includes the Hohara ALS focus. Four of the individuals had a moderate number of neurofibrillary tangles in the locus coeruleus, substantia nigra, raphe nucleus, periaqueductal grey and hippocampus in addition to the histological changes of ALS. All four came from the vicinity of Hohara; symptoms of ALS developed in 1979, 1987, 1991 and 1993. Two had family history of ALS, and one, of parkinsonism-dementia. These findings confirm that Kii type ALS occurs continuously in and near the Hohara focus’’ [41].
Virginia Bozzoni et al., “Physical activity and football: Physical activity and soccer have been postulated as risk factors for ALS, ever since an Italian prosecutor, Raffaele Guariniello, ordered an inquiry to investigate the main causes of death in soccer players. This inquiry was set up in response to the complaint of a football coach, worried about the use of illegal drugs among players. In a large retrospective cohort study, Belli and Vanacore (2005) considered 24,000 professional soccer players who had played, in the period 1960–1996, in Italy’s 3 top leagues (A, B and C). They found a surprisingly high prevalence of ALS: eight cases among the entire cohort, giving a SPMR (standardized proportionate mortality ratio) 1 of 11.58.
Subsequently, several epidemiological studies attempted to confirm these findings.
Chio et al. (2005), performed a retrospective incidence study in a rigorously defined cohort of Italian professional soccer players, playing in Serie A and Serie B in the period 1970–2001. They found a total of five ALS cases during the 137,078 person-years follow-up (the number of expected cases for this period was 0.77); the consequent SMR was 6.5 (95% CI, 2.1–15.1), revealing a significant relationship between ALS and professional soccer activity. A dose-response relationship between the duration of activity and ALS risk was observed. In a subsequent article, the follow-up was extended to include the period 2001–2006, and another 3 ALS cases were found (SMR 6.45; 95% CI 2.70–12.70; p<0.00001).the authors compared ALS incidence in soccer players with that recorded basketball players and cyclists: the fact that they did not observe ALS cases in the latter two cohorts strengthened the soccer/ALS hypothesis.
Subsequently, similar studies conducted in National Football League players in the United States confirmed an apparent increased risk of neurodegenerative disorders, in particular ALS, in this cohort. Nevertheless, a retrospective case-control study published in 2012 failed to confirm the association.
The supposed relationship between football and ALS is also supported by several case-report studies.
Valenti et al. (2005), reported a case-control study conducted to assess the trustworthiness of this association: 300 ALS cases and an equal number of matched controls were enrolled, and exposure data concerning physical activity were collected through a questionnaire. The results did not show a role for sports, particularly soccer, as risk factors for ALS.
A similar case-control study (Huisman et al., 2013) also failed to detect a statistically significant association.
In a recent article, Hamidou et al. (2014), reported a literature review conducted according to the Meta-analysis of Observational Studies in Epidemiology guidelines, in which the authors considered data drawn from six databases (PubMed, SCOPUS, Science Direct, IngentaConnect, Refdoc, Cochrane Database): of the 37 papers included, two (5.5%) provided class I evidence and five (13.5%) class II evidence. The others offered class III (21.6%), IV (43.2%) and V (16.2%) evidence. The results were stratified according to the type of exposure: i) occupation, ii) soccer and American football, iii) physical activity related to sport and work, iv) proxies of physical activity (PA).
They found nine studies relating to soccer and American football: two class V, one class IV, four class III, one class II and one class I. They concluded that football may be considered a possible risk factor for ALS (level C evidence).
A literature review by Armon (2007) did not confirm the excess of ALS in Italian soccer players (level C evidence, two class III articles; the evidence level of the conclusions is based on re-analysis of the data, rather than on the class and level of the original methodology).
Possible risk factors in football players
The cause of this supposed association is not well-defined, but several hypotheses have been postulated to explain the epidemiological findings that seem to indicate its existence. Football players may be exposed to various risk factors that could contribute to neurodegenerative processes, namely excessive PA, repeated head injuries, exposure to pesticides and dietary supplements or illegal substances.
Heavy physical activity
It has been supposed that heavy physical stress could enhance the production of reactive oxygen species, leading to nucleic acid damage (Julien, 2001), particularly in association with dietary supplements (branched-chain amino acids, BCAAs) and drugs.
Several epidemiological investigations have been conducted on the basis of this assumption.
Veldink et al. (2005), in their case-control study denied any association between PA and motor neuron disease risk.
Huisman et al. (2013), found a weak association between leisure time PA and ALS (OR 1.08, 95% CI 1.02–1.14). “The lack of association with occupational PA and the absence of a dose-response relationship” led them to affirm that “not PA per se but rather a genetic profile or lifestyle promoting fitness increases ALS susceptibility”.
Pupillo et al. (2014), recently published a European population-based case-control study, whose results allowed them to assert that PA is not a risk factor and may be considered protective against the disease.
In a recent literature review of six databases (PubMed, SCOPUS, Science Direct, IngentaConnect, Refdoc, Cochrane Database), Hamidou et al. (2014), stated that PA is not a risk factor for ALS (level A evidence).
Repeated head injuries
The possibility of a link between head trauma and ALS risk has been analyzed repeatedly over more than a century. Retrospective case-control studies often offer only equivocal results, due to selection bias or because of imprecise definition of trauma, its gravity and temporal relationship with ALS onset.
This hypothesized link was examined in a meta-analysis study (Chen et al., 2007), which showed a moderately elevated ALS risk associated with a history of head injuries (OR 1.7; 95% CI 1.3–2.2).
Armon (2007) stated that head trauma is probably not a risk factor for ALS (level B evidence).
A European population-based case-control study (Pupillo et al., 2014) evaluated several environmental risk factors for ALS, including head trauma, and denied any association with motor neuron disease.
Exposure to pesticides and dietary supplements/illegal substances.
Pesticides are widely employed on soccer and football fields. As previously seen (pesticide paragraph), pesticide exposure is involved in triggering neurodegenerative diseases, particularly in predisposed individuals. No epidemiological study has shown direct involvement of pesticides in increasing.
Virginia Bozzoni et al., ALS among soccer and football players.
Exposure to food toxicants or to illegal substances used to enhance physical performance have been considered possible risk factors since the first study (Belli and Vanacore, 2005). Creatine monohydrate, growth hormone (GH) and BCAAs are the dietary supplements that have been the main focus of attention. Creatine monohydrate supplements, often used by athletes, were initially thought to be capable of worsening motor neuron function, but experimental results disproved this hypothesis. Conversely, oral administration of creatine monohydrate in murine models has been shown to improve motor neuron performances and demonstrates oxidative stress reduction.
In a multicenter double-blind study 107 ALS patients were randomized to receive either daily creatine monohydrate (5g/day) or placebo and were followed up for nine months, during which the authors monitored their clinical status and ALS functional rating scale scores. The results showed that creatine monohydrate did not significantly improve motor, respiratory or functional capacity. A trend toward improved survival in patients was noticed, as affirmed in another published report.
In a phase I study, Atassi et al. (2010), remarked that creatine monohydrate has been shown to be able to cross the blood-brain barrier. Indeed oral administration of creatine monohydrate, 15g, to six patients recruited in the study was associated with increased in vivo brain creatine concentrations and decreased glutamate concentrations.
Growth hormone is often taken by athletes, alone or in association with other anabolic steroids, to increase muscle mass and strength. GH was not found to be dangerous for motor neuron survival; rather, it showed a trophic effect on the nervous system. GH deficiency in ALS patients has been reported in the literature (Saccà et al., 2012; Pellecchia et al., 2010).
A deficiency in GH secretion similar to that seen in human ALS has been reported in hSOD1 (G39A) transgenic mice; a reduction in the expression of the IGF-1 receptor α-subunit in skeletal muscle and lumbar spinal cords suggested impaired signaling within these tissues.
A recent clinical trial failed to detect clinical and survival improvement in ALS patients administered GH.
Branched-chain amino acids (leucine, isolucine and valine) are widely used among athletes to stimulate muscle protein synthesis, improve physical resistance and reduce delayed onset muscular soreness. BCAAs were initially evaluated as a possible therapeutic option for ALS patients but the results obtained were discouraging.
They have therefore been investigated as risk factors for ALS. Piscopo et al. (2011) examined the effect of a diet enriched with BCAAs in C57BI/6J mice and demonstrated that BCAAs are able to down-regulate the expression of some antioxidant genes and can alter oxidative stress pathways in the brain; the authors concluded that caution should be exercised in the use of these dietary supplements by athletes.
Venerosi et al. (2011) demonstrated that BCAA supplements can exacerbate motor deficits in G39A mice. Electrophysiological tests on mouse brain slices showed impaired synaptic function, but no increased glutamate toxicity. Although these results do not confirm a role for BCAAs in ALS etiology in mice models, they indicate a complex effect on the central nervous system, depending on genetic background, and suggest that they should be used with caution.
Physical activity and football: conclusions
The literature in this field shows that an increased ALS incidence may be present only among soccer players, while professionals from other sports do not demonstrate a higher risk for ALS. Analysis of single risk factors for soccer players has given negative results. A potentiating effect of a number of risk factors acting together cannot be excluded in soccer players. Studies on the effect PA in the general population show that it is not a risk factor and it may be considered protective against the disease’’ [45].
Yu Yu et al., “Radiation/electromagnetic Fields.
Radiation has been considered as a potential risk factor for ALS since a myeloradiculopathy presentation can be caused by electrical injuries with a long latency period. We saw an association with ALS in women who had an exposure to occupational radiation 10 to 30 years earlier; the very large OR, the small number of exposure cases (n = 10/2 for cases/controls), and gender effect specificity suggest that results may not be meaningful.3 studies previously reported associations between radiation or electromagnetic field exposure; thus, further investigation of such exposures is warranted. Radiation has been investigated in many case-control studies, again with inconsistent results. Electrical-related occupations (OR = 1.3, 95% CI = 1.1–1.6), and exposure to electromagnetic fields (OR = 2.3, 95% CI = 1.29–4.09) were associated with ALS. In a cohort mortality study at five large United States electric utility companies (139,905 men), mortality from ALS was associated with the duration of work in jobs with electromagnetic field exposure (RR = 2.0, 95% CI = 1.0–9.8). A prospective case-control study in Denmark of utility workers (n = 30,631, 81% male), did not find significant linkages between electromagnetic field exposure and neurological diseases’’ [46].
Kuzuhara S, “The Muro district includes the southern coastal mountainous areas of the Kii peninsula of Japan where high incidence foci of ALS and parkinsonism-dementia complex (PDC) exist. “Muro disease” refers to the endemic ALS in Muro, and the oldest description is in a book published in 1689. Miura reported high prevalence of ALS in Muro in 1911, and the first epidemiological survey by Kimura and Yase in 1960s disclosed extremely high prevalence of ALS in Hohara and Kozagawa. The high incidence was, reported to have disappeared by early 1980s as in Guam. In 1990s we resurveyed and found not only continuous high ALS incidence but also neuropathologically-verified PDC in Hohara. ALS and PDC here frequently affected one individual simultaneously and members in a family. Neuropathological changes were common to ALS and PDC, showing a combination of ALS changes and many neurofibrillar tangles (NFTs) in the brainstem and cerebral cortex, suggesting ALS/PDC may be a single entity with different clinical manifestations, “ALS-parkinsonism-dementia complex”. TDP-43-positive inclusions were confirmed in all cases examined. Age-adjusted incidence rates during 1950 and 2000 have showed that incidence of ALS gradually declined for 50 years while that of PDC rose up steeply in 1990s, suggesting changing pattern of ALS/PDC that had occurred in Guam in 1970s. Continuing high incidence of ALS/PDC and high familial occurrence suggest that primary cause of Kii ALS/PDC may be genetic rather than environmental” [47].
Sara Zarei et al., (ALS) is a late-onset fatal neurodegenerative disease affecting motor neurons with an incidence of about 1/100,000. Most ALS cases are sporadic, but 5–10% of the cases are familial ALS. Both sporadic and familial ALS (FALS) are associated with degeneration of cortical and spinal motor neurons. The etiology of ALS remains unknown. Mutations of superoxide dismutase 1 have been known as the most common cause of FALS. In this study, we provide a comprehensive review of ALS. We cover all aspects of the disease including epidemiology, comorbidities, environmental risk factor, molecular mechanism, genetic factors, symptoms, diagnostic, treatment, and even the available supplement and management of ALS. This will provide the reader with an advantage of receiving a broad range of information about the disease.
“(ALS) is a fatal motor neuron disorder that is, characterized by progressive loss of the upper and lower motor neurons (LMNs) at the spinal or bulbar level.
ALS was first described in 1869 by French neurologist J.-M. Charcot. The disease became well known in the United States when baseball player Lou Gehrig was diagnosed with the disease in 1939. ALS is also known as Charcot disease in honor of the first person to describe the disease, Jean-Martin Charcot, and motor neuron disease (MND) as it is one of the five MNDs that affect motor neurons. There are four other known MNDs: Primary lateral sclerosis (PLS), progressive muscular atrophy (PMA), progressive bulbar palsy (PBP), and pseudobulbar palsy.
ALS is categorized in two forms. The most common form is sporadic (90–95%) which has no obvious genetically inherited component. The remaining 5–10% of the cases are familial-type ALS (FALS) due to their associated genetic dominant inheritance factor. The first onset of symptoms is usually between the ages of 50 and 65. The most common symptoms that appear in both types of ALS are muscle weakness, twitching, and cramping, which eventually can lead to the impairment of muscles. In the most advanced stages, ALS patients will develop symptoms of dyspnea and dysphagia.
Most of the reviews about ALS focus on a specific area of the diseases such as molecular mechanism, treatment, diagnostic, etc. This review will attempt to provide an up-to-date overview of all aspects of ALS. It will first cover the epidemiology and comorbidities of the disease, followed by known environmental risk factors such as smoking, chemical exposure, and radiation.
Improving the understanding of ALS pathogenesis is critical in developing earlier diagnostic methods as well as proposing new effective treatments. Thus, this review will present the most recent studies related to molecular mechanisms, genetics, ALS symptoms, diagnostic examinations, and treatments. Furthermore, due to the fact that there has been only one Food and Drug Administration (FDA) approved drug for ALS treatment, this review will also address nutritional supplements, as well as respiratory and nutritional managements that help alleviating the symptoms. This comprehensive study will inevitably lead to the better understanding of ALS and assist in extending the life expectancy associated with ALS by establishing a basis of knowledge that can be used to improve care.
During 1990’s, the number of reported cases of ALS was between 1.5 and 2.7 per 100,000 in Europe and North America. Recent studies have shown that disease prevalence has not increased over the past decade, as the incidence rate remains at 2.7/100,000 (95% confidence interval [CI] 2.63–2.91 and as of 2008, the prevalence of ALS was 0.32/100,000 (95% CI 9.78–10.86). Multiple studies have shown an increased risk in the male to female ratio (male: female =1.5:1), although other studies have shown a balance in this ratio.
The mean age of onset of ALS varies from 50 to 65 years with the median age of onset of 64 years old. Only 5% of the cases have an onset <30 years of age. ALS incidence is most pronounced in people 80 years or older (10.2/100,00 in men; 6.1/100,000 in women). One hypothesis for the increased incidence in older population is related to the variation in care for these patients.
Although most cases of ALS are sporadic, about 5% of the cases have a family history. The age of onset for FALS is about a decade earlier than for sporadic cases. There is also geographical loci form of ALS where prevalence is 50–100 times higher in certain locations than in any other part of the world. These population include parts of Japan, Guam, Kii Peninsula of Japan, and South West New Guinea. Although this evidence is not concrete, it is believed that the increased incidence of ALS in these regions is due to environmental factors, specifically a neurotoxic nonprotein amino acid, β–methylamino-L-alanine (BMAA) in the seeds of the cycad Cycas micronesica produced by a symbiotic cyanobacteria in the roots of the cycad that are commonly found in these areas. It is hypothesized that patients in these regions who develop ALS have an inability in preventing BMAA accumulation. More research is needed in South America to corroborate data for the rest of the continent, yet new studies show that the incidence of sporadic ALS in Uruguay is similar to those found within North America and Europe. Thus, it is important to continue epidemiologic studies of ALS in areas where little work has been done to identify vulnerable populations within Africa, Central America, and South America.
A possible relationship between ALS and sports participation has been proposed but not demonstrated. In a cohort study of Italian professional football players, a severe increase in the incidence of ALS was found. This study, conducted by the National Institute of Environmental Health Science, showed a correlation between head injuries in football players and an increased risk of ALS (odds ratio [OR] =3.2; 95% CI =1.2–8.1). Traumas to other parts of the body were not associated with increased risk.
Analysis of co-morbidities of ALS
Retrospective cohort studies have shown that the incidence in certain concomitant diseases and comorbidities is significantly different in ALS-affected population in comparison to the general population. German cohort studies examining comorbidities prior to diagnosis found that while cardiovascular risk factors were the most common comorbidities in ALS patients (31.5% vs. 40%), they still had a significantly higher incidence in the general population than in their affected cohort. The discrepancy in incidence could suggest that ALS causes a protective mechanism for cardiac disorders or that there are genetic benefits for those predisposed to the disorder. ALS patients had a significantly lower incidence of arterial hypertension (31.5% vs. 47.2%), coronary heart disease (8.6% vs. 9.3%), myocardial infarctions (6.4% vs. 7.0%), diabetes mellitus (7.2% vs. 10.6%), and hypercholesterolemia (17.9% vs. 65.6%). These results were echoed when results were stratified by various age groups to prevent confounding. With regards to comparing ALS types and their onset time to patient comorbidities, only a difference in hypercholesterolemia incidence was seen when comparing bulbar onset to spinal onset (23.6% vs. 15.7%; P < 0.05). This shows only a slight association with how cardiovascular comorbidities can alter ALS progression.
Of the diseases most likely to be found in ALS patients prior to their diagnosis, a higher incidence in neurological disorders was noted when compared to the general population. The increased incidence of neurological diseases in ALS patients may be indicative of similar underlying genetic factors between ALS and other neurological disorders. A history of depression (22.8% vs. 11.6%), dementia (5.8% vs. 1.3%), Parkinson’s (1.8% versus 0.1–0.2%), and epilepsy (1.6% versus 0.45–1%) was found more frequently within the study cohort than with the rest of the population. The results were echoed in other studies that have indicated a greater likelihood of ALS development and progression when examining patients with a high rate of psychological disorders.
Development of depression is one of the most common secondary symptoms associated with ALS. Previous studies have reported a prevalence of depression of 4–56% depending on the assessment measure. Depression has a negative effect on the quality of life of patients with ALS. In a recent study, 131 patients with ALS were evaluated to estimate the prevalence of depression. The results showed 29% prevalence of mild depression and 6% prevalence of severe depression. In this study, more than one-third of ALS patients were receiving antidepressant to treat sialorrhea, pseudobulbar affect, and insomnia, which may explain the lower rates of severe depression in ALS patients compared to the prevalence of depression in the general population (10%). Physical impairment and duration of the disease did not predict depression in the cohort which suggests that depression is not related to advanced ALS or approaching end of life. ALS-related symptoms, including cramps, stiffness, shortness of breath, swallowing difficulty, insomnia, loss of appetite, increased saliva, uncontrolled laughing or crying, fasciculations, anxiety, fatigue, and pain, did not appear to contribute to depression with the exception of anxiety which is a known symptom of depression. Other studies have also shown that, even in late stages of ALS, major depression is rare.
When comparing the survival rates of patients with mental health comorbidities, it was noted that patients suffering from Parkinson’s and ALS had a slower progression of their disease and a greater survival time (P = 0.03). It may be possible that the pathological development of Parkinson’s disease (PD) could slow down the effects of ALS onset by altering the speed of nervous degeneration and disease progression. It is also possible that the administration of L-DOPA or the decreased levels of endogenous dopamine could offer an antioxidant protective effect in those susceptible to ALS, similar to that seen in multiple studies comparing ALS incidence and Vitamin E consumption. Greater evidence will be necessary to confirm correlations and association between these comorbidities and ALS incidence.
Radiation/electromagnetic fields
Laboratory studies have demonstrated that in vitro exposures to extremely lowfrequency electromagnetic waves generate a bigger quantity of cellular reactive oxygen than normal this oxidative damage can lead to ALS since oxidative stress has a role in ALS pathogenesis. Studies have observed that electromagnetic fields cause DNA strands to break in brain cells, leading to cell death (apoptosis and necrosis).Such reaction could be the reason for the association between electromagnetic fields and ALS risk. None of the current studies found a conclusive connection among electromagnetic field exposure, oxidative stress in neurons, and/or ALS development.
Diet
Previous studies state that consuming high level of glutamate and fat can have adverse effects on ALS patients while Omega 3 fatty acids, Vitamin E, and fiber can have defensive impact. According to previous studies, overstimulation of glutamate receptors leads to high intracellular calcium levels, which can initiate selective neuron death similar to ALS mechanism. Glutamate is found in protein-rich foods, tomatoes, mushrooms, milk, and cheese. Normally glutamate does not cross the blood-brain barrier, hence it is not known if dietary glutamate affects neurotransmission. There are areas of the brain called circumventricular organs, which are susceptible to plasma glutamate levels Omega 3 has been known to possess anti-inflammatory characteristics, which in turn would theoretically reduce inflammation caused by neuronal death. Omega 3 in conjunction with Vitamin E has been reported to reduce ALS risks up to 60%. These nutrients appear to act together in a summative way.
A large number of the information regarding environmental factors are based off questionnaires, all of which rely on subjects’ memories, leading to recall bias. Because of this, there may be a lack of information about the frequency and the amount of exposure to environmental factors. Also, this may also lead to the absence of biological markers in order to validate patient claims of exposure or pinpoint the possible action site.
Due to ALS prolonged onset, it is difficult to isolate an exact environmental factor. In order to identify or narrow down possible ALS risk factors, a cohort study utilizing mice as a control and experimental group could be appropriate. Starting out with an emphasis on the most sought out factors (smoking, heavy metals, physical activity, diet, radiation, and chemical exposure). In order to track changes accurately, a type of biomarker specific to the possible risk factor could be designed, to theoretically track the progression of the disease.
Finding the molecular mechanisms by which motor neurons degenerate in ALS will aid in better understanding the disease’s progress. Also, elucidation of molecular mechanisms can yield insight into developing strategies for newer treatments. The molecular basis of ALS is an intriguing issue that warrants in-depth research and investigation.
The most common cause of ALS is a mutation of the gene encoding the antioxidant enzyme superoxide dismutase 1 (SOD1). Mutant SOD1 has a structural instability that causes a misfold in the mutated enzyme, which can lead to aggregation in the motor neurons within the central nervous system (CNS). Several hypotheses have been proposed in regards to the mechanism underlying the mode of action of mutant SOD and the subsequent neurodegeneration seen in ALS. The most important proposed hypothesis for the pathogenesis of ALS includes glutamate excitotoxicity structural and functional abnormalities of mitochondria, impaired axonal structure or transport defects, and free radical-mediated oxidative stress. Even though these mechanisms play a critical role in neurodegeneration, they all are considered as secondary events in the causes behind ALS onset.
Glutamate excitotoxicity: Glutamate is synthesized in the presynaptic terminal. Uptake of glutamate into synaptic vesicles is facilitated by vesicular glutamate transporters. During a normal neurotransmission process, glutamate is released into the synaptic cleft, where it activates postsynaptic receptors. Upon release of the vesicle, glutamate is removed from the synaptic cleft by several glial and neuronal cell transporter proteins, such as excitatory amino acid transporters (EAATs). This continuous release and removal of glutamate maintain a concentration gradient balance and avoid the induction of excitotoxic neuronal damage.
The motor cortex and spinal cord of ALS patients and transgenic SOD1 mouse model were found to have reduced astroglial glutamate transporter EAAT2, which leads to increased extracellular glutamate, over-stimulation of glutamate receptors, and excitotoxic neuronal degeneration. Furthermore, this causes an excessive influx of calcium, excessive firing of motor neurons, and initiation of several destructive biochemical processes within the cell, which are all known as important pathophysiological processes in familial and sporadic forms of ALS. Glutamate excitotoxicity contributes to the neurodegeneration either through activation of Ca2+-dependent enzymatic pathways by increasing the influx of Na+ and Ca2+ ions or by the generation of free radicals.
Aberrant EAAT2 messenger RNAs (mRNAs) were found in neuro-pathologically affected areas and cerebrospinal fluid (CSF) of ALS patients. These abnormalities included intron-retention and exon-skipping. According to these findings, aberrant mRNA is the main reason for the decrease in EAAT2 receptors among ALS patient.
Structural and functional abnormalities of mitochondria: Mitochondrial dysfunction also plays an important role in the motor neuron degeneration. Mitochondria are membrane bound organelles that have a significant role in vital processes such as intracellular energy production, cellular respiration, calcium homeostasis, and control of apoptosis. Accumulating evidence suggests that abnormalities in mitochondrial morphology and biochemistry contribute to the pathogenesis of ALS. Functional defects and altered mitochondrial morphology such as fragmented network, swelling, and augmented cristae were found in soma and proximal axons of skeletal muscle and spinal motor neurons of ALS patients.
In the spinal cords of ALS patients, mutant SOD1 is deposited on the cytoplasmic face of the outer membrane and matrix of mitochondria. The increase of misfolded mutant SOD1 in spinal cord mitochondria is considered as the main reason for mitochondrial dysfunction that leads to abnormal functioning of ATP production, calcium homeostasis, axonal transport of mitochondria, and apoptotic triggering.
Mitochondria act as the powerhouse of every cell by converting energy into ATP that is, essential for the metabolism of the cells. Disturbed energy homeostasis and ATP deficits have been reported in the skeletal muscle biopsies of ALS patients. The normal process of electron transport chains is perturbed by the presence of mutant SOD1, causing less production of ATP. Some studies have demonstrated a decreased activity of respiratory chain complexes I and IV that are associated with defective energy metabolism.
In addition to energy homeostasis, another major function of mitochondria in neurons regards buffering cytosolic calcium levels. Thus, unraveling the relationship between aberrant mitochondria, calcium dysregulation, and neuronal death is critical for the understanding of ALS pathogenesis. Calcium is one of the most significant intracellular messengers that play an important role in the regulation of metabolic pathways, neuronal development, and synaptic transmission. Mutant SOD1 has been found to disrupt calcium homeostasis. Several studies have shown that intracellular calcium is misregulated in ALS patients. A lower cytosolic Ca2+ buffering ability has been found as a principal risk factor for motor neuron damage.
Several studies reported the loss of Ca2+ binding proteins such as calbindin-D28K and parvalbumin in the motor neurons of ALS patients. These findings are in agreement that with studies showing neurons lost early in the ALS development have low cytosolic Ca2+ buffering capabilities due to the loss of Ca2+ binding proteins. Meanwhile increasing the cytosolic Ca2+ buffering capacity has shown to reduce motor neuron degeneration.
Regulating mitochondrial transport along axons is an essential task for the survival of neurons due to the mitochondria’s key role in ATP generation, calcium buffering, and apoptotic signaling. Mitochondria are constantly being transported and docked at the same time in areas with high demand of ATP and calcium homeostasis such as growth cones, nodes of Ranvier, and synaptic terminals. Thus, any defects in mitochondrial transport will lead to energy depletion and disruption in Ca2+ buffering, activating synaptic dysfunction and a loss of neurons. Several laboratories have identified disrupted axonal transport of mitochondria in ALS patients. The axonal transport alteration impairs the degradation and recycling process of abnormal mitochondria, thus increasing the amount of dysfunctional mitochondria at distal axons, mitochondrial movement can also be suppressed in both anterograde and retrograde directions.
Increased mitochondrial transport may slow axonal degeneration by delivering healthy mitochondria to axons while removing the damaged one from distal synapses. Mitochondria have been an attractive target for ALS therapy development and drugs, such as olexisome, are already in clinical trial for ALS patients.
Finally, mutant SOD1 aggregates may also interfere with components of mitochondrial-dependent apoptotic machinery, such as B-cell lymphoma 2 (Bcl-2), which is a regulator protein that controls cell death. Thus, causing the stimulation of premature apoptotic cascade activation is leading to the release of cytochrome C in the presence of Bcl-2, which directly contributes to neuromuscular degeneration and neuronal dysfunction.
Impaired axonal structure and transport defects
Motor neurons are highly polarized cells with long axons that can be more than a meter in length and are thus vulnerable to damage. In addition to transmitting nerve impulses axons also transport organelles, RNA, proteins, lipids, and other cell parts to the axonal compartments. Moving toward the soma is called retrograde and is performed by cytoplasmic dynein molecular motors while moving toward the synaptic structures at the neuromuscular junction is an anterograde transport and is conducted by microtubule-dependent kinesin.
Axonal transport in ALS patients is compromised. Dysregulation of axonal transport and the axonal compartment play a critical role in the pathophysiology of ALS. In several experiment with mutant SOD1 mice, loss of neurotrophic signaling and defective axonal transport were observed early in the disease process. Both anterograde and retrograde transport were impaired by the presence of mutant SOD1.
Several pathways may be responsible for the impaired axonal transport in cases with mutant SOD1. Some of the most important mechanisms involve defective mitochondrial function or energy depletion, disruption of kinesin function by tumor necrosis factor, and excitotoxic damage by glutamate. Defective axonal transport causes an accumulation of neurofilaments, mitochondria, and autophagosomes in degenerated motor neurons. This leads to further hindrance of axonal transport and eventual motor neuron death.
Reactive oxygen species (ROS) or free radicals form as natural byproducts of the normal metabolism of oxygen. ROS accumulation causes severe damages to cell structures. The term oxidative stress is used to define a disturbance in the balance between the production ROS and cell’s antioxidant defenses [94]. Losing the ability to detoxify the harmful reactive intermediates will lead to cell demise.
Increased oxidative damage has been reported in ALS case biopsies and altered redox reactions were among the earliest theories of how mutant SOD1 could cause cytotoxicity.
SOD1 is a major antioxidant protein, thus a mutation in this gene could cause cytotoxicity. Elevation of free radicals and increased oxidative damage were found in CSF, serum, and urine samples of ALS patients. In addition, oxidative damage to RNA species was found in both mutant SOD1 mouse models, as well as in human CNS biopsies.
Sporadic ALS accounts for the majority of the cases of ALS, but genetic causes have been known to play a role. FALS occurs due to mutations in specific genetic loci. The inheritance follows a clear Mendelian pattern and is primarily autosomal dominant.
The clinical and pathological presentation of FALS and sporadic ALS are similar. Genetic testing can be used to differentiate inherited versus sporadic ALS and also to rule out other diseases that clinically mimic ALS. Starting with the discovery of mutations in the SOD1 gene, which codes for copper/zinc ion-binding SOD, 18 other genes have been identified in association with FALS. The additional genes that are known to cause FALS include: TARDBP, encodes TAR DNA-binding protein 43 (TDP-43); FUS, which codes for fusion in sarcoma; ANG, which codes for angiogenin, ribonuclease, and the RNAase A family 5; OPTN, which codes for optineurin; and C9orf72.
SOD1 mutations, which account for 20% of cases of FALS and 5% of SALS, cause cytotoxicity, which still has an unclear pathophysiology. TARDBP mutations represent 5–10% of FALS mutations. TDP-43 and FUS, which represents 5% of FALS mutations, are part of the process of gene expression and regulation including transcription, RNA splicing, transport, and translation, as well as processing small regulatory RNAs. ANG, responsible for the remaining 1% of FALS, is a gene, coding for an angiogenic factor that responds to hypoxia. OPTN is a gene involved in open-angle glaucoma, where a mutation in this gene eradicates the inhibition of nuclear factor kappa-beta activation, changing the distribution of OPTN in the cytoplasm. Approximately, 50–60% of FALS patients have mutations arising from the 19 genes that have been identified to date. SOD1 and C9orf72 mutations most often cause FALS, but their rates vary across population.
FALS is inherited at a rate of 5–10% for all cases of ALS where family history of the disease is known In the United States, a founder effect has been identified for the A4V mutations in SOD1, whereas in Europe, this mutation is uncommon. OPTN mutations occur most often in Japanese population. To date, there has been no evidence for geographic variability in FUS and TARDBP. As soon as more causative genes are identified in FALS, mutation frequencies across different FALS population will be available. The lifetime risk of ALS is 1:450 for women and 1:350 for men. As family size increases, there is a greater likelihood of two family members having SALS.
Close to 50% of FALS cases can be attributed to specific genes, and most are seemingly rare, highly penetrant, de novo mutations within affected families. Genome-wide association studies (GWAS) has allowed for the identification of common variables that are coupled to this disease. Another technique is next-generation sequencing (NGS), otherwise known as massively parallel sequencing, which provides a way to map mutations for single gene diseases. Together, GWAS and NGS have helped to identify genetic variables that are seen in parallel with a higher risk for developing ALS. Ascertaining accurate clinical phenotypes is essential for the success of these techniques to avoid false positive results.
Family aggregation studies for SALS patients have shown that many people who have common neurodegenerative disorders also have ALS, possibly indicating the presence of a susceptible gene that could be responsible for increasing neurodegeneration in kindreds.
Many GWAS for SALS have resulted in identifying genes that are associated to the ALS disease. Two new susceptible loci, 19p13.3 (UNC13A) and 9p21.2, were identified through collaborative research that combined study pools to elucidate effectively both genes.[146,303] If more research groups work on collaborative efforts, it is highly probably that more molecular pathways and genetic markers could be identified” [48].
Ludolph AC et al., “Lathyrism is a toxic disease of the motor system constantly associated with primary consumption of the seed of Lathyrus sativus (chickling or grass pea). Neurological examination was performed on 38 affected subjects drawn from four regions of the Indian subcontinent endemic for lathyrism. All showed a consistent and largely symmetric pattern of neurological deficit similar to that displayed by 5 Europeans with longstanding lathyrism now living in Israel. Hallmarks of lathyrism include a pyramidal pattern of motor weakness combined with greatly increased tone in the thigh extensors and adductors and in the gastrocnemius muscles so that the more severely affected walk on the balls of their feet with a lurching scissoring gait. Extensor plantar responses are uniformly present in such cases, and the knee and ankle tendon reflexes are exaggerated and often clonic. Hoffmann signs and exaggerated biceps and/or triceps tendon jerks are also found in the most severely affected. Sensory signs are absent, although perverse sensations in the legs are frequently reported at the onset. Walking difficulties commonly begin suddenly but may also appear subacutely or insidiously. Some individuals experience partly reversible symptoms suggestive of a diffuse CNS excitation of somatic, motor and autonomic function” [49].
Arjun L. Khandare et al., “Neurolathyrism is a non progressive motor neuron disorder engendered by the prolonged over-consumption of Lathyrus sativus (grass pea) seeds which contain a neurotoxic amino acid, β-N oxalyl- L-α, β-diaminopropionic acid (β-ODAP). It is characterized by spastic paraparesis in the hind limbs. The present study was conducted in 105 households (HHs) of Gondia district in Maharashtra, India, where grass pea is cultivated and consumed to assess the health implication of its consumption.
A cross-sectional survey was carried out in 105 HHS in five villages and grass pea samples were collected for β-ODAP estimation. Amino acid analysis was also done, neurolathyrism cases were identified by snowball sampling method and neurological examination was carried out.
The study revealed that 61 per cent of population was consuming this pulse as a part of diet. β-ODAP concentration in grass pea was high in Bora village (1254.5 ± 528.21 mg %) and less in Malgaon village (413.6±415.79 mg %). The nutritional status of the people was within the normal range (BMI 18± 3.40 kg/m2) in the surveyed households. Consumption of grass pea was observed to be less than 25g.
The cases of neurolathyrism declined in all the studied villages due to reduced β-ODAP exposure through Lathyrus sativus consumption, the grass pea was cultivated and consumed in Gondia district of Maharashtra State.
Lathyrus sativus (grass pea/khesari dal) is a high yielding; drought-tolerant legume crop containing 31 per cent protein, 41 per cent carbohydrate, 17 per cent total dietary fiber. The protein content of grass pea seeds is higher compared to other legume seeds3. Grass pea is cultivated and consumed in India, Nepal, Bangladesh and in many parts of Africa that are prone to recurrent droughts. It is often considered a lifesaver crop or as insurance crop. It is an abundant source of protein for human and animal consumption. Presence of homoarginine (a source of potent vasodilator nitric oxide) in grass pea can be used to explore its inherent potential to treat cardiovascular disorders associated with vascular endothelial function.
The major grass pea growing States in India are Bihar, Madhya Pradesh, Maharashtra, West Bengal and Chhattisgarh. Other States of Uttar Pradesh, Rajasthan, Gujarat, Andhra Pradesh and Karnataka produce this pulse in a limited quantity. People from lower economic sections consumed this legume crop as a staple food during famines and floods.
Excessive consumption of grass pea containing β -N oxalyl- L-α, β-diaminopropionic acid (β-ODAP) 9, analogue of glutamate for 2-3 months as a monotonous diet, leads to an irreversible motor neuron disorder, neurolathyrism. Pathological symptoms are caused by symmetrical degeneration of pyramidal tracts in the spinal cord11, and loss of pyramidal cells in the motor cortex of the brain leads to the impairment of the hind limb movements. Reports suggest that β-ODAP is metabolized in human beings. Metabolic pathway of β-ODAP in human beings is not exclusively known. Earlier epidemiological studies have depicted illiteracy, poverty and young age as major risk factors for neurolathyrism.
The harmful effects depend on quantity of the pulse consumed, its β-ODAP content, method of cooking; and nutritional status of the individual. Susceptibility to neurolathyrism varies from person to person even within a family.
The National Institute of Nutrition (NIN), Hyderabad, Andhra Pradesh, India, has undertaken this study to assess the grass pea cultivation and its health implications in Gondia district of Maharashtra State in India with the following objectives. (i) quantity of the pulse consumed and exposure of β-ODAP per day, (ii) its health implications, (iii) nutritional status of the consumers, and (iv) to determine the level of β-ODAP in the grass pea samples’’ [50].
Uccelli R et al., “ (ALS) is a rare and devastating neurological disorder of the adult age with a prognosis of about 2-3 years from the onset of the disease. No clear cause has been identified but it seems to be a multifactorial disease with genetic and environmental components involved. Increments of mortality rates were observed since 1980 both in Italy and in many other countries. The objective of the present study is to describe the distribution of ALS mortality in Italy in the period 1980-2001 detecting single municipalities or clusters with high mortality levels for motor neuron disease (MND). ALS represents the main part (85%) of the MND group which is globally identified by the IX ICD (International Classification of Diseases and Causes of Death) 335.2 code. Death numbers and standardized mortality ratios (SMR) for MND were calculated for all Italian municipalities through the ENEA mortality database system (data source: National Institute of Statistics-ISTAT), using national mortality rates as reference. Subsequently, in order to detect municipal clusters, spatial analysis was performed. Out of the 8,099 Italian municipalities, 132 where characterized by SMR values higher than expected. 16 clusters with significant high relative risk values (RR) were identified, 12 out of them including only a single municipality. Only 22 of the municipalities with high SMR were included in the clusters. In conclusion, the two different epidemiological methodologies demonstrated to be widely complementary in detecting the geographical distribution of the disease in terms of risk for populations. A first selection of the priority areas where analytical studies should be carried on, in order to identify risk factors associated to ALS, is tentatively suggested’’ [51].
Schwartz GG et al., “We recently reported that U.S. mortality rates for motor neuron disease (MND) at the level of the state are associated with well water use. Data at the state level may not accurately reflect data at the individual level. We therefore examined the association between MND mortality and well water use utilizing data from smaller geographic units that may better reflect exposure and disease at the individual level.
We used data on age-adjusted MND mortality rates at the level of the county, obtained from the CDC, and corresponding data on the prevalence of well water use, obtained from the U.S. Geological Survey. Data were analyzed by multivariate linear regression and by Getis-Ord Gi*, a measure of spatial clustering.
Age-adjusted mortality rates for MND in 923 U.S. counties were significantly correlated with the prevalence of well water (p
These findings support the hypothesis that an agent present in well water plays an etiologic role in ALS. Further study of water use among individuals with ALS is warranted’’ [52].
Li W et al., “The role of human endogenous retroviruses (HERVs) in disease pathogenesis is unclear. We show that HERV-K is activated in a subpopulation of patients with sporadic (ALS) and that its envelope (env) protein may contribute to neurodegeneration. The virus was expressed in cortical and spinal neurons of ALS patients, but not in neurons from control healthy individuals. Expression of HERV-K or its env protein in human neurons caused retraction and beading of neurites. Transgenic animals expressing the env gene developed progressive motor dysfunction accompanied by selective loss of volume of the motor cortex, decreased synaptic activity in pyramidal neurons, dendritic spine abnormalities, nucleolar dysfunction, and DNA damage. Injury to anterior horn cells in the spinal cord was manifested by muscle atrophy and pathological changes consistent with nerve fiber denervation and reinnervation. Expression of HERV-K was regulated by TAR (trans-activation responsive) DNA binding protein 43, which binds to the long terminal repeat region of the virus. Thus, HERV-K expression within neurons of patients with ALS may contribute to neurodegeneration and disease pathogenesis [53].
Chio A et al., “We previously found an increased risk for ALS in Italian professional soccer players actively engaged between 1970 and 2001 (n =7325). The present study extends previous work with a prospective follow-up of the original cohort to 2006 and investigates the risk of ALS in two other cohorts of professional athletes, basketball players (n =1973) and road cyclists (n =1701). Standardized morbidity ratios (SMRs) were calculated. Among soccer players three new cases of ALS were identified, reaching a total of eight ALS cases (mean age of onset, 41.6 years). The number of expected cases was 1.24, with an SMR of 6.45 (95% CI 2.78-12.70; p<0.00001). The risk of ALS was higher for careers lasting >5 years, for midfielders, and for players engaged after 1980. No basketball player and no cyclist developed ALS. This prospective extension of the Italian soccer player’s cohort survey confirms the highly significant risk of developing ALS, the young age of onset, the dose-effect risk and a predilection for midfielders. The absence of ALS cases in professional road cyclists and basketball players indicates that ALS is not related to physical activity per se” [54].
Chiò A et al., “The cause of (ALS) is still unknown. A possible relationship between ALS and sport participation has been supposed, but never definitely demonstrated. We studied a cohort of 7325 male professional football players engaged by a football team from the Italian First or Second Division in the period 1970-2001. ALS cases were identified using different concurrent sources. Standardized morbidity ratios (SMRs) were calculated. During the 137,078 person-years of follow-up, five ALS cases were identified (mean age of onset, 43.4 years).3 cases had a bulbar onset, significantly more than expected (P = 0.003). Since the number of expected cases was 0.77, the overall SMR was 6.5 [95% confidence interval (CI), 2.1-15.1]. The SMR was significantly increased for an ALS onset before 49 years, but not for older subjects. A significant increase of the SMR was found in the periods 1980-1989 and 1990-2001, whereas no ALS case was found in the 1970-1979 period. A dose-response relationship between the duration of professional football activity and the risk of ALS was found (>5 years, 15.2, 95% CI, 3.1-44.4; < or =5 years, 3.5, 95% CI, 0.4-12.7). Our findings seem to indicate that playing professional football is a strong risk factor for ALS’’ [55].
Belli S et al., “The objective of the study is to investigate the mortality experience of Italian soccer players and to discuss the findings in the light of possible long term effects of doping.
Standardized proportionate mortality ratio (SPMR) and standardized proportionate cancer mortality ratio (SPCMR) were computed for 350 deceased subjects deriving from a list of about 24,000 active Italian soccer players from 1960 to 1996 in the 3 top leagues.
When considering SPMRs, there is a substantial adherence of observed to expected mortality, with the only exception of mortality for diseases of the nervous system (13 obs. vs. 6 exp.) mainly explained by an excess of ALS (8 obs. vs 0.69 exp.). As far as SPCMRs are concerned, some digestive cancers (namely: colon cancer, liver cancer and pancreas cancer) show a doubled risk.
A high risk for ALS is observed among Italian soccer players. Epidemiological data on association between sport and (ALS) are contrasting. On the basis of the overall available evidence we suggest a possible connection between dietary supplements or drugs used to enhance sporting performance and ALS pathogenesis. Further epidemiological studies are needed to confirm these specific mortality risks among soccer players” [56].
Aurélie Delzor et al., “(ALS) is the most common motor neurone disease. It occurs in 2 forms: familial cases, for which several genes have been identified and sporadic cases, for which various hypotheses have been formulated. The β-N-methylamino-L-alanine (L-BMAA) toxin has been postulated to be involved in the occurrence of sporadic ALS. The objective of the French BMAALS programme is to study the putative link between L-BMAA and ALS.
The programme covers the period from 1 Jan. 2003 to 31 Dec. 2011. Using multiple sources of ascertainment, all the incident ALS cases diagnosed during this period in the area under study (10 counties spread over 3 French regions) were collected. The standardized incidence ratio will be calculated for each municipality under concern. By applying spatial clustering techniques, over incidence and under incidence zones of ALS will be sought. A case–control study, in the subpopulation living in the identified areas, will gather information about patients’ occupations, leisure activities and lifestyle habits in order to assess potential risk factors to which they are or have been exposed. Specimens of drinking water, food and biological material (brain tissue) will be examined to assess the presence of L-BMAA in the environment and tissues of ALS cases and controls. This part aims to highlight differences in life habits between patients SALS and controls. Criteria for selecting patients with ALS are as follows: familial history cases are excluded; last known address must be in an over incidence or under incidence area and if possible, vicinity with other affected people, which may suggest a close source of an environmental risk factor leading to ALS. Controls will be matched on age at diagnosis, sex and city and should not present any neurological pathologies. Chosen patients and controls will be submitted to a semistructured interview, for example, systematic questions with the possibility of free interview to look in more depth at particular issues raised. The questionnaire has been developed by the consortium specifically for the BMAALS programme.
On the basis of the initial results of clustering,26 a number of clusters to investigate was selected a priori: 3 over incidence areas in Limousin, 2 in Languedoc-Roussillon and 4 in Rhône-Alpes; with an expected number of 4 patients in each cluster (and 4 controls), this will represent a total of about 72 interviews. The same number of interviews for patients and controls will be performed in under incidence areas. Owing to the short survival time of the disease, the number of living patients diagnosed between 2003 and 2011 is low.
Cyanobacterial and L-BMAA hypotheses are tested via questions about: drinking water; bathing habits; food consumption including dietary supplements (to be specified if applicable), the type of supplement is informed and irrigation water, if any. The aim of the questionnaire is to obtain a comprehensive description of patients’ habits in all aspects of their lives. Hence, it will be made clear that questions are not just about the time immediately preceding the diagnosis.
To assess exposure to cyanotoxins indirectly, an ad hoc questionnaire is a useful supplement to direct collection of environmental samples Hence, samples will be taken in case and control environments to test for the presence of cyanobacteria in water (the same probe as described above) and for further chemical analysis (in water and food)” [57].
Gilberto M. Manzano et al: The classification arrived at its present version: in mammals, the motor nerves are composed of Aα, Aβ, Aγ, Aδ and C fibers and pure sensory nerves are composed of the Aβ, Aδ and C components in mammals, the motor nerves are com-posed of Aα, Aβ, Aγ, Aδ and c fibers and pure sensory nerves are composed of the Aβ, Aδ and c components.
“There are variations between species in relation to the spectrum of fibers constituents of nerves and therefore the border between the different fiber groups are arbitrary20 and shows some variations21. Considering these, we may quote the classification given by an important textbook on human neuroanatomy11 where Aα fibers are described as having sizes between 12-22 μm and velocities from 70 to 120 m/s, Aβ from 5-12 μm and velocities 30-70 m/s, Aγ 2-8 μm and velocities 15-30 m/s, Aδ 1-5 μm and velocities 5-30 m/s, B<3 μm and velocities 3–15 m/s and C 0.1-1.3 μm and velocities 0.6–2.0 m/s’’ [58].
Mireille Be´ langer et al., “The energy requirements of the brain are very high, and tight regulatory mechanisms operate to ensure adequate spatial and temporal delivery of energy substrates in register with neuronal activity. Astrocytes—a type of glial cell—have emerged as active players in brain energy delivery, production, utilization, and storage. Our understanding of neuroenergetics is rapidly evolving from a ‘‘neurocentric’’ view to a more integrated picture involving an intense cooperativity between astrocytes and neurons. This review focuses on the cellular aspects of brain energy metabolism, with a particular emphasis on the metabolic interactions between neurons and astrocytes” [60].
Bertamini M et al., “The mnd mouse spontaneously develops slowly evolving motoneuron pathology leading to progressive motor impairment. There is strong evidence that a complex interplay between oxidative stress, mitochondria abnormalities and alteration of glutamate neurotransmission plays an important role in the pathogenesis of motor neuron diseases. Therefore, we investigated the presence of mitochondrial dysfunction in frontal, central (comprising the motor area) and occipital regions of the cerebral cortex and in the spinal cord of 35-week-old mnd mice. Lipid peroxide derivatives reacting with thiobarbituric acid (TBARS) were measured in the cervical, thoracic and lumbar spinal cord. In addition biochemical and behavioural analyses were carried out in mnd mice chronically treated with l-carnitine from the 11th to the 34th week of life (mndT mice). Slight but significant alterations of mitochondrial enzyme activities were seen in the mnd cortical regions. The central area was the most affected and both complex I, IV and citrate synthase were decreased with respect to controls. The rate of oxygen consumption (QO2) was markedly decreased in both the upper (cervical + upper portion of the thoracic region) and lower (lumbar + lower portion of the thoracic region) mnd spinal cord. The level of TBARS showed a rostro-caudal trend to increase, being 30% higher in the lumbar tract of mnd mice in comparison with controls. L-carnitine treatment increased the mitochondrial enzyme activities in cortical regions towards control value and was effective in enhancing QO2 and decreasing TBARS levels in the spinal cord of mndT. Behavioural testing showed that L-carnitine significantly delayed the onset of motor behaviour impairment. This beneficial effect was declining at 35 week of age, when the biochemical measurements were performed [62].
Tesfaye W et al., “(ALS) is a complex multi-pathogenic neurodegenerative disorder primarily characterized by the loss of upper motor neurons in the brain and lower motor neurons in the spinal cord and the brain stem. As a result of this neuronal loss, motor function declines progressively which then leads to impairments in respiratory function and eventually death within 2–5 years of diagnosis The exact mechanisms underlying the selective loss of motor neurons in the disease are not yet known. However, several mechanisms appear to contribute, including glutamate excitotoxicity oxidative stress protein aggregation cytoskeletal and axonal transport abnormalities, inflammation hyperexcitability and altered energy metabolism. It is believed that several of these mechanisms are interconnected and together are responsible for the progression of the disease. To date, there is only one approved medication available to treat ALS, namely riluzole, which only increases survival by 3 months. Therefore, it is imperative to look for other potential treatments that can halt the progression of the disease and extend life. In this review, we discuss impairments in energy metabolism in ALS and review the effects of metabolic therapies investigated so far in animal models of ALS and/or patients with ALS.
Energy metabolism disturbances in ALS
There are many lines of evidence for the role of disturbed energy metabolism in ALS pathogenesis. Energy homeostasis, the balance between energy intake and expenditure, appears to be compromised in patients with ALS. Besides, some patients exhibit hypermetabolism (an increased metabolism at rest) body weight loss and abnormal metabolism of lipids), which are likely to contribute to disease. These and many studies discussed below reveal problems in energy metabolism in whole tissues and even the whole organism. It is not entirely clear to which extent the metabolic changes found in ALS patients and ALS models only arise as a result of neurodegeneration. Moreover, little is known about the specific metabolic changes within the motor neurons themselves and if they contribute to the degeneration. Other early reviews discuss reasons for the specific degeneration of motor neurons in ALS.
Specific dysfunctions in energy producing pathways in ALS
Under normal physiological conditions, glucose is the main energy substrate for neuronal tissue and a major fuel for muscles. However, there are indications of defects in the glucose metabolizing pathways in ALS namely in the glycolysis and the TCA cycle pathways in CNS and muscle (Palamiuc et al., 2015; Tefera et al., 2016). In addition, there is decreased glucose uptake in numerous brain regions and spinal cord of animal models and patients with ALS. In muscle, namely the tibialis anterior muscle of the superoxide dismutase 1 (SOD1G86R) mice, the activities of the enzyme phosphofructokinase 1 (PFK1), the rate limiting enzyme in glycolysis, were reduced. Furthermore, in the gastrocnemius muscle of SOD1G93A mice, the activities of the TCA cycle enzyme 2-oxoglutarate dehydrogenase (OGDH) were found to be reduced in the late symptomatic phases of the disease, indicating impairments in the TCA cycle pathway. Furthermore, changes in the expression of several genes involved in energy metabolism have been observed in muscle, brain and spinal cord tissues and cells isolated from them. Particularly, genes involved in the mitochondrial electron transport chain are significantly altered. Mitochondria are the main site of energy production and they are also the main sites for the generation of reactive oxygen species. Compromised mitochondrial function has been recognized for a long time in many neurodegenerative disorders, which is expected to lead to reduced production of ATP and increased oxidative stress. Similarly in ALS, numerous studies have shown functional and structural abnormalities in mitochondria that give rise to reduced oxidative phosphorylation and subsequent decreased generation of ATP. In addition, in vitro studies showed that mitochondrial dysfunction in astrocytes may lead to generation of free radicals and release of toxic factors that could contribute to motor neuron dysfunction. In conclusion, pathological changes in the energy metabolizing pathways that lead to deficiency in ATP production and/or utilization may contribute to progression of the disease.
Role of glial cells in metabolism and ALS
Although ALS is mainly characterized by the selective loss of motor neurons, neighboring non-neuronal cells such as astrocytes, microglia and oligodendrocytes can contribute to the progression of the disease. The metabolic coupling between neurons and astrocytes has long been known. During neurotransmission, neurons release glutamate into the synapse which can be taken up by astrocytes. It is then metabolized to glutamine, which can be supplied to neurons to resynthesize glutamate. Many immunohistochemical studies have shown the loss of glutamate transporters in astrocytes in spinal cord and brain of patients with ALS Deficiencies in glutamate uptake and the glutamate-glutamine cycle can lead to problems in neuronal signaling and promotes excitotoxicity). according to the astrocyte-neuron lactate shuttle hypothesis, glutamate stimulated metabolism of glucose in astrocytes produces lactate which can be transferred to neurons to be utilized as fuel Altered gene expression of the astrocytic lactate efflux transporter (monocarboxylate transporter 4, SLC16A4) together with reduced lactate levels found in the spinal cord of hSOD1G93A mice further suggest that metabolic interaction between neurons and astrocytes may be disrupted in ALS).
The potential of metabolic coupling between neurons and oligodendrocytes has gained a lot of interest in recent years and it appears that oligodendrocytes also can release lactate. Studies displayed the involvement of oligodendrocytes in the pathogenesis of ALS Healthy oligodendrocytes can metabolically support neurons by providing energy metabolites via monocarboxylate transporters). In contrast, the transport of glycolytic substrates such as lactate from oligodendrocytes to neurons is found to be disrupted in SOD1G93A mice spinal cord Consistent with this, the expressions of the oligodendrocytic lactate transporter monocarboxylate transporter 1 (SLC16A1) was decreased in the spinal cords of patients with ALS and SOD1G93A mice suggesting abnormalities in lactate transfer.
Microglia have been shown to be involved in ALS pathogenesis). Microglia are immune cells within the CNS, similar to macrophages, which can either have a neuroprotective or a neurotoxic role during ALS progression. It is believed that microglia protect neurons early in the disease stage. However, as the disease progresses, activated microglia can be pro-inflammatory and neurotoxic In vivo imaging studies demonstrated increased microglia activation and inflammatory activity in spinal cord during symptomatic stages in the SOD1G93A mouse model reduced expression of mutant SOD1 in microglia delayed late disease progression and increased survival of SOD1G37R mice.
In conclusion, perturbations in glial cells can affect neurons and directly or indirectly can contribute to energy deficits in neurons. The studies discussed demonstrate the importance of non-neuronal cells in supporting neurons in various ways and indicate that abnormalities in glial cells can exacerbate the progression of the disease.
High calorie diets in ALS
A number of studies have tried to correct the energy deficit using metabolic therapies in animal models and patients with ALS, with the hope that an improved metabolism can delay symptoms and extend survival in ALS A study by Dupuis et al. (2004) in SOD1G86R mice found aberrations of energy metabolism such as reduction of adiposity, increased energy expenditure and lipolysis, and examined the effects of a high fat diet that consisted of regular chow supplemented with 21% (wt/wt) butter fat and 0.15% (wt/wt) cholesterol. A 20% increase in the mean survival of these mice compared to mice fed with control diet was demonstrated and motor neuron loss was reduced. Moreover, SOD1G93A mice fed either a high fat diet (containing 47% fat, 38% carbohydrates and 15% protein) or with control diet (constituting 17% fats, 64% carbohydrates and 19% protein) starting at 6 weeks of age also showed an increase in survival and the onset of motor symptoms and weight loss were delayed significantly. While mice treated with control diet died by 180 days, high fat died treated mice survived until 220 days and some of them survived more than 270 days. On a similar note, several studies have alluded that body mass index is strongly correlated with survival of patients with ALS. A prospective study of over a million people for about 14–28 years demonstrated that obese people have a 30–40% lower risk of developing ALS compared to those with a healthy weight. Given the strong link found between low body mass index and decreased survival with ALS, a randomized double blind phase 2 clinical study was performed in small number of patients with ALS to test the safety and tolerability of high caloric diets, which were either high in carbohydrates or high in fat. Compared to an isocaloric control diet, the high carbohydrate-high calorie diet was safe, tolerable and effective in delaying weight loss and prolonging survival in the patients. Further large scale trials are needed to corroborate these promising effects. Also due to the small sample size, a beneficial effect of a high fat high calorie diet cannot yet be ruled out completely. On the other hand, high fat diets are linked with side effects such as brain inflammation, cognitive deficits, and anxiety and depressive behaviors.Together, these studies show the importance of adequate energy supply to slow the progression of ALS” [63].
Belli S et al., “The objective of the study is to investigate the mortality experience of Italian soccer players and to discuss the findings in the light of possible long term effects of doping.
Standardized proportionate mortality ratio (SPMR) and standardized proportionate cancer mortality ratio (SPCMR) were computed for 350 deceased subjects deriving from a list of about 24,000 active Italian soccer players from 1960 to 1996 in the 3 top leagues (A, B and C).
When considering SPMRs, there is a substantial adherence of observed to expected mortality, with the only exception of mortality for diseases of the nervous system (13 obs. vs. 6 exp.) mainly explained by an excess of ALS (8 obs. vs 0.69 exp.). As far as SPCMRs are concerned, some digestive cancers (namely: colon cancer, liver cancer and pancreas cancer) show a doubled risk.
A high risk for ALS is observed among Italian soccer players. Epidemiological data on association between sport and (ALS) are contrasting. On the basis of the overall available evidence we suggest a possible connection between dietary supplements or drugs used to enhance sporting performance and ALS pathogenesis. Further epidemiological studies are needed to confirm these specific mortality risks among soccer players’’ [64].
Malek AM et al., “Exposure to pesticides and agricultural chemicals has been linked to (ALS) although findings have been inconsistent. A meta-analysis of studies published through May, 2011 was conducted to investigate the association of pesticide exposure and risk of ALS.
Six peer-reviewed studies that met criteria were included in a meta-analysis of men involving 1,517 ALS deaths from one retrospective cohort study and 589 ALS or motor neuron disease cases from five case-control studies. A random effects model was used to calculate sex-specific pooled odds ratios (ORs).
Evidence was found for an association of exposure to pesticides and risk of ALS in male cases compared to controls (OR=1.88, 95% CI: 1.36-2.61), although the chemical or class of pesticide was not specified by the majority of studies.
This meta-analysis supports the relationship of exposure to pesticides and development of ALS among male cases compared to controls. The weight of evidence links pesticide exposure to ALS; however, additional prospective studies with a target exposure group are necessary to better elucidate the relationship. Future research should focus on more accurate exposure assessment and the use of job exposure matrices’’ [65].
Nina Feddermann‐Demont et al., “(ALS) has been reported to occur with increased incidence amongst physically active people. The role of extrinsic risk factors as physical activity, head trauma and drug/pesticide‐exposure in the pathophysiology of ALS and especially in the context of practising sports remains controversial.
We retrospectively studied exposure to extrinsic factors in 92 ALS patients in the presymptomatic stage. Metabolic equivalents (METs) were calculated and the association of physical activity, drug intake, head trauma and participation in specific sports (football [soccer], ice hockey) with age at symptom‐onset was evaluated.
95 percent of patients considered themselves physically active before symptom‐onset. Total MET‐values varied broadly and there was no correlation between vigorous physical activity and age at symptom‐onset. Mild traumatic brain injury (prevalence = 16.7%) was the most frequent diagnosis after head/neck injury. A history of ≥1 head/neck injuries was associated with a younger age at symptom‐onset (61.8 ± 11.0 vs. 54.1 ± 13.0, p =.013). In former football and ice hockey players the rate of vigorous physical activities was increased (p <.05), whereas total MET‐values, frequency of head injuries and analgesic intake were not different compared to other ALS patients.
History of head injuries was the only extrinsic risk factor associated with accelerated neurodegeneration in ALS. There was no evidence for extrinsic factors predisposing former football and ice hockey players to ALS. Our data therefore support the hypothesis that not increased physical activity per se, but other unknown environmental factors and/or genetic profile or lifestyle‐promoting physical fitness increases ALS susceptibility’’ [67].
M.I. Vargas et al., “Ischemia of the spinal cord is a rare entity with a poor prognosis. Brain ischemia is no longer a diagnostic challenge; on the contrary, ischemia of the spinal cord remains difficult, particularly in children. In this article, we illustrate the principal causes in children and adults, clinical presentation, different techniques for the diagnosis by MR imaging (diffusion, spinal MR angiography, and 1.5 versus 3T), pathophysiology, and differential diagnosis. We will discuss current knowledge, perspectives, and pitfalls.
Incomplete syndrome of the spinal artery syndrome
Ischemia may be localized at the level of the anterior horns; in this case, clinical presentations may be the following:
1) Acute paraplegia (pseudopoliomyelitic form) without sensory abnormalities and without sphincter dysfunction.
2) Painful bilateral brachial diplegia in the case of a cervical lesion (the man-in-the-barrel syndrome).
Progressive distal amyotrophy due to chronic lesions of the anterior horns; this form may be misdiagnosed as lateral amyotrophic sclerosis” [68].
Seongkyun Kim et al., “Determining the fundamental architectural design of complex nervous systems will lead to significant medical and technological advances. Yet it remains unclear how nervous systems evolved highly efficient networks with near optimal sharing of pathways that yet produce multiple distinct behaviors to reach the organism’s goals. To determine this, the nematode roundworm Caenorhabditis elegans is an attractive model system.
We investigated the vulnerability of the Caenorhabditis elegans connectome by performing computational experiments that (a) “attacked” 279 individual neurons and 2,990 weighted synaptic connections (composed of 6,393 chemical synapses and 890 electrical junctions) and (b) quantified the effects of each removal on global network properties that influence information processing. The analysis identified 12 critical neurons and 29 critical synapses for establishing fundamental network properties. These critical constituents were found to be control elements—i.e., those with the most influence over multiple underlying pathways.
Additionally, the critical synapses formed into circuit-level pathways. These pathways provide evidence for the importance of backward locomotion, avoidance behavior, and social feeding behavior to the organism; the potential roles of specific neurons whose functions have been unclear; and both parallel and serial design elements in the connectome—i.e., specific evidence for a mixed architectural design.
Function derives from form, and with unrelenting natural selection, nervous systems must achieve complexity as efficiently as possible. Network analysis provides a normative approach to analyze the solution, and vulnerability analysis helps provide a more experimental and less assumption-laden analysis of individual component contributions to information processing.
In this study, we tested the effects of attacks on every neuron and synaptic connection in the C. elegans connectome to identify and characterize the most critical constituents of the network.
To our knowledge, this is the first study to analyze network robustness for individual node and edge attacks on an entire nervous system. We identified 12 neurons and 29 synapses critical for clustering, information integration and propagation.
Although one might have expected the clustering, efficiency, and betweenness values for individual neurons and synapses to be the most important factors determining these same properties at the network level, control structures—i.e., those that influence multiple others—prove the most important. From an evolutionary-development (evo-devo) perspective, the additions of higher-order control structures can produce the largest effects on information processing, providing the most ‘bang for the buck’. Thus, to most affect clustering, new neurons should project paired synapses to neighbors. To most affect global efficiency, new neurons or synapses should be placed in central positions that shorten the largest number of prior existing pathways. These more centralized neurons not only contribute to overall efficiency and information integration, they also become traffic centers, and thereby have greater influence (and thus control) in the connectome. Finally, to most affect network traffic flow (i.e., global betweenness), new neurons or synapses should provide new pathways that decrease a larger number of path lengths and should also normally link to other control structures, leading to the formation of chains of control units (i.e., node1!node2!node3).
Not only were there critical control structures at the cellular level (neurons and synapses), the synapses organized into larger control structures at a higher circuit level—into full or partial circuits. The vulnerability analysis seemed to uncover fractal-like control in the network. Yet these network control systems must translate to circuits that accomplish the organism’s biological goals, and there was in fact a close correspondence between the identified critical pathways and biological function. The AVA-based pathway is a large component of the backward locomotor circuitry. Identifying this circuitry as a control pathway with respect to its relative influence in the network suggests that there is high volume use of this circuitry. This in turn suggests that backward locomotion likely has multiple trigger stimuli and is utilized by Vulnerability-Based Critical Constituents of C. elegans Connectome PLOS Computational multiple biological functions that require maneuverability. Indeed, given the apparent need for the worm to reverse prior to changing directions (for omega turns), the finding may highlight the significance of backward movement for navigation more generally. At the same time, to the extent that backward movement reflects the avoidance of particular trigger stimuli, it might also point to the importance of avoidance behavior.
For the PVP-based pathway, given the sensory to motor structure, as well as the AQR neuron being implicated in social feeding and the VD01 neuron in locomotor control, our analysis appears to implicate a social-feeding circuit as a second major critical pathway in the connectome.
Moreover, the pathway result implicates PVP, DVC, and AVH involvement in social feeding—neurons whose complete functional roles in information processing in the adult network remain unclear. The third pathway segment has also been implicated in avoidance behavior.
Taken together, our results suggest that the network topology is particularly organized around backward movement, avoidance behavior and social feeding, suggesting a primary significance of these functions to the organism. Indeed, backward movement is likely required for maneuverability in multiple behavioral functions. Thus, the C. elegans appears to have evolved under strong selection pressures for maneuverability, to avoid undesirable and harmful circumstances and to negotiate social interactions. The significance of social behavior would highlight the fundamental importance of behavioral strategies in animals to overcome inevitable competition for resources.
With respect to architectural design, the AVA-based pathway has 3 general components:
A second-layer interneuron (RIB), command neurons (AVE, AVA, PVC), and motor neurons. It is a subcircuit for information integration, sensorimotor integration, motor control, and motor performance. Beginning with a second layer interneuron (RIB), it suggests that multiple prior circuit segments may use this critical pathway to produce backward locomotion: for multiple avoidance stimuli to tap into this (backward locomotion) circuit segment. This finding is comparable to Gray et al.’s suggestion of a “common substrate” foraging subcircuit. Such subcircuit structures lend support for a serial-based architectural design structure (i.e., input! central processing! output).
Our second PVP-based pathway contained a sensory to motor structure that suggests a more complete behavioral circuit, and itself provides evidence for separable behavioral circuits, and thus a parallel design structure. Our findings point to a mixed or hybrid architectural design. studies of the C. elegans have appeared to find evidence for this mixture—with modules aligning both with known biological function to some degree (supporting parallelism), as well as with the input-output serial design structure to some degree (e.g., sensory and first-layer interneurons dominating one module, second-layer interneurons another, and command interneurons and motor neurons another). Our study highlights and extends these findings by identifying specific neurons, synapses and pathways using a vulnerability approach that implicates specific circuit structures as having critical roles in the connectome. Our findings may also extend those by others who have found significant neural activity across a large extent of the nervous system during activities such as locomotion. Even when such global processing occurs, there is likely a hierarchy of importance or influence among the network components, which can be characterized.
Our findings derive from theoretical analyses, they require empirical verification, especially to test the hypotheses generated from our study: most notably, the functional roles of PVP (sensorimotor integration), DVC (information integration, motor control), and AVH (information integration, motor control) in social feeding behavior (as well as the other possible roles of DVC and AVH derived in the S1 Text).
The nematode C. elegans is a valuable model system to identify potentially fundamental design elements of nervous systems. Further specification of these principles in the C. elegans and other nervous systems, including that of humans, will lead to the ultimate goals of a true understanding of nervous system design, and the subsequent medical and technological advances that follow [70].
To further examine the properties of the critical neurons, we first compared all critical neurons to the noncritical ones on multiple network factors using the Mann-Whitney U test (Fig 2B). The critical neurons have higher degree (D), strength (Str), average weight (AW), nodal efficiency (E(i)), and nodal betweenness centrality (B(i)) values than the noncritical neurons. Only the nodal clustering coefficient (C(i)) showed no differences between the critical and noncritical groups.
Thus, as a group, the critical neurons were more connected, more strongly connected, more closely connected to others in the network (i.e., the shortest average path lengths), and generally positioned as important control centers regarding the number of shortest pathways passing through them and thus the number of pathways influenced by them. These results support others that have shown the significance of nodal network properties such as degree and betweenness centrality in the connectome.
Multiple factors distinguish the critical neurons, but it is important to clarify their impact on each vulnerability measure VC,E,B individually. One might assume that each vulnerability measure would be most affected by its corresponding nodal property: e.g., individual neuron values for clustering coefficient (C(i)) determining global VC. This is not what we found—in fact in no case was the same local nodal property the leading factor influencing global network vulnerability. To evaluate the relationship of global VC,E,B to nodal network properties of the intact network, we examined correlations of D, Str, AW, C(i), E(i), B(i) to VC,E,B without taking the absolute values. The S1 Text contains a detailed discussion of the results. Neurons most critical for network clustering (VC) send dual (or more) projections to other neurons that project to each other, producing the largest clusters. Those most critical for global efficiency (VE) generally have the most influence—or control—in the network, regarding the number of pathways sharing them and thus the number influenced by them. The critical VE neurons do not necessarily have high individual efficiency values, but they do normally have high individual betweenness values. And those critical for network betweenness (VB) also appeared to be those with most control in the network, but the effect is more contextual—it depends on the other possible available routes when the node is lost (detours), the loss in nodal betweenness centrality of the attacked node, and the changes generated in others due to the loss of the attacked connections. Most critical neurons for VB also tended to be connected to other nodes with high nodal betweenness centrality, creating a linked control structure” [70].
Property (Clustering: C, Efficiency: E, and Betweenness: B) induced by 279 single neuronal attacks. Mean vulnerability value for 279 neurons are indicated by the solid black lines; black dashed lines indicate the threshold value (mean plus 3SDs). (D) Depiction of the 12 critical neurons with their connections to each other along the anterior-posterior body axis in the C. elegans body context. The network property for each neuron is shown (C, E, and B) with the critical grade in parenthesis; thickness of links indicates the weight grade of a link [70] (Figure 17).
Figure 17: Vulnerability results for each network property and a visualization of critical neurons. (A-C) Sorted vulnerability values (red) for each network.
Vulnerability
“We quantified vulnerability, V, to determine the effect of the loss of a neuron or synapse on global information processing, as measured by 3 key graph-theoretic structural properties: the clustering coefficient (VC), global efficiency (VE), and the average betweenness centrality of the network (VB). The vulnerability of a network to a particular loss might also be considered the degree of lethality or redundancy, or the functionality enabled by a component’s addition” [70].
List of 12 critical neurons. (S=SENSITIVE, I=INTERNEURON,M =MOTORNEURON) Neuron VC VE VB Functional role [5] Birth Time (min) AQR (S) 3σ Aerotaxis (O2 and CO2), regulating social feeding and bordering behavior (i.e., aggregation in densest part of bacterial lawn), and suppressing innate immunity 1280.
AVAL (I) 10σ 9σ 3σ backward locomotion 312
AVAR (I) 11σ 7σ backward locomotion 311
AVER (I) 3σ backward locomotion 329
AVHL (I) 4σ Unknown yet 347
DA01 (M) 3σ backward locomotion 309
DVA (I) 3σ Mechanosensory integration; Sensory-motor integration during locomotion 296
DVC (I) 5σ backward locomotion 351
PVCL (I) 3σ forward locomotion modulating response to harsh touch to tail 449
PVCR (I) 3σ forward locomotion modulating response to harsh touch to tail 450
PVPR (I) 4σ Ventral cord pioneering (first during development for axon guidance) 310
VD01 (M) 3σ Sinusoidal body movement 1460 [70].
Lee BC et al., “Among the putative mechanisms, by which extremely low frequency (ELF) magnetic field (MF) may affect biological systems is that of increasing free radical life span in organisms. To test this hypothesis, we investigated whether ELF (60 Hz) MF can modulate antioxidant system in mouse brain by detecting chemiluminescence and measuring superoxide dismutase (SOD) activity in homogenates of the organ. Compared to sham exposed control group, lucigenin-initiated chemiluminescence in exposed group was not significantly increased. However, lucigenin-amplified t-butyl hydroperoxide (TBHP)-initiated brain homogenates chemiluminescence, was significantly increased in mouse exposed to 60 Hz, MF, 12 G for 3 h compared to sham exposed group. We also measured SOD activity that plays a critical role of the antioxidant defensive system in brain. In the group exposed to 60 Hz, MF, 12 G for 3 h, brain SOD activity was significantly increased. These results suggest that 60 Hz, MF could deteriorate antioxidant defensive system by reactive oxygen species (ROS), other than superoxide radicals. Further studies are needed to identify the kind of ROS generated by the exposure to 60 Hz, MF and elucidate how MF can affect biological system in connection with oxidative stress [71].
Piazza O et al., “Trauma has long been hypothesized but never proven to be a risk factor for amyotrophic lateral sclerosis (ALS). This hypothesis may now have a renaissance due to recent reports in the lay press on ‘the Italian motoneuron mystery’, i.e. the disclosure of 33 diagnosed ALS cases in a subpopulation of 24000 soccer players of the top three Italian divisions from the 1960s to 1996. Could the repetitive brain trauma that soccer players experience for controlling and advancing the ball with their heads represent an environmental risk factor for developing ALS in genetically predisposed individuals? By critically reviewing the scarce literature and ‘surrounding evidence’ (Medline, CDC, lay press, Italian health officials), we have looked for a potential relationship between (1) soccer and head trauma and (2) head trauma and subsequent development of amyotrophic lateral sclerosis. Whereas the brain traumatizing effect of soccer seems to be out of the question, the findings of the few retrospective studies on ALS and neurotrauma are conflicting. Taken together, however, the literature would still support the concept of soccer, head trauma, and ALS being interrelated, with high levels of athleticism/physical activity perhaps playing an additive part. To further clarify this issue, extensive prospective epidemiological investigations on ALS following neurotrauma as well as carefully designed animal studies will have to be conducted” [73].
Scotter EL et al., “Therapeutic options for patients with amyotrophic lateral sclerosis (ALS) are currently limited. However, recent studies show that almost all cases of ALS, as well as tau-negative front temporal dementia (FTD), share a common neuropathology characterized by the deposition of TAR-DNA binding protein (TDP)-43-positive protein inclusions, offering an attractive target for the design and testing of novel therapeutics. Here we demonstrate how diverse environmental stressors linked to stress granule formation, as well as mutations in genes encoding RNA processing proteins and protein degradation adaptors, initiate ALS pathogenesis via TDP-43. We review the progressive development of TDP-43 proteinopathy from cytoplasmic mislocalization and misfolding through to macroaggregation and the addition of phosphate and ubiquitin moieties. Drawing from cellular and animal studies, we explore the feasibility of therapeutics that act at each point in pathogenesis, from mitigating genetic risk using antisense oligonucleotides to modulating TDP-43 proteinopathy itself using small molecule activators of autophagy, the ubiquitin-proteasome system, or the chaperone network. We present the case that preventing the misfolding of TDP-43 and/or enhancing its clearance represents the most important target for effectively treating ALS and front temporal dementia” [74].
Patrick Sweeney et al., “A hallmark of neurodegenerative proteinopathies is the formation of misfolded protein aggregates that cause cellular toxicity and contribute to cellular proteostatic collapse.
Misfolded proteins exist in cells together with unfolded,intermediately folded, and correctly folded species. In healthy cells, misfolded proteins are either degraded or refolded correctly by chaperone proteins that are involved in protein folding and trafficking as well as intermediate stabilization. It is now believed that many, if not all, proteins can form amyloid fibrils under appropriate biochemical conditions. Many disease-associated amyloidogenic proteins have extensive regions of intrinsic disorder in their free soluble forms and have specific, often short, internal aminoacid sequences that are necessary and sufficient to support aggregation. These same motifs can be found in other non-disease proteins, and when liberated from rest of protein these fragments will aggregate into cytotoxic amyloid fibrils.
Once formed, higher order amyloid aggregates are highly resistant to degradation. Proteasomes can degrade only single chain polypeptides, and also require the proteins to be partially or fully (in the case of proteasomes) unfolded. The amyloid state is extremely stable thermodynamically, because of the extensive contacts made between the protein chains of the polymer. The thermodynamic stability of amyloid aggregates also contributes to their ability to convert native proteins into amyloid forms (i.e., to seed prion-like propagation CLIPs (chaperones linked to protein synthesis) are physically associated with the translational machinery and oversee quality control of newly translated proteins.
CLIPs comprise a large family of proteins, and evidence suggests that different CLIPs associate with different classes of proteins. The second set of chaperones, heat shock proteins (HSPs), protects the proteome from denaturing environmental stressors, including thermal, oxidative, and hypoxic stresses. CLIPs and HSPs have different modes of transcriptional regulation in yeast: CLIPs are generally down-regulated under conditions of proteotoxic stress, whereas HSPs are up-regulated.
Sub networks of differentially regulated chaperones and co-chaperones have also been identified in C. elegans and in the human brain. it has been found that as the human brain ages, a subset of chaperones consisting primarily of CLIPs are repressed, and chaperones that help protect the proteome against misfolded protein toxicity are induced mimicking proteotoxic stress; these differences are even more pronounced in the brains of people with Alzheimer’s, Huntington’s, or Parkinson’s disease. Misfolded proteins that are not immediately refolded are actively sequestered in spatially and functionally segregated quality control compartments. In yeast, the juxtanuclear quality control (JUNQ) compartment concentrates soluble misfolded proteins that are either later refolded by chaperones or degraded by the ubiquitin proteasome system (UPS). The insoluble protein deposit (IPOD) compartment, which may be equivalent to the aggresomes found in mammalian cells, sequesters insoluble aggregates. The sequestration of aggregated misfolded proteins may in many cases serve a beneficial role – by preventing misfolded proteins from saturating chaperones and proteasomes, facilitating their clearance via the UPS or through autophagy, or by preserving them for subsequent refolding and return to use in the cell” [75] (Figures 18-25).
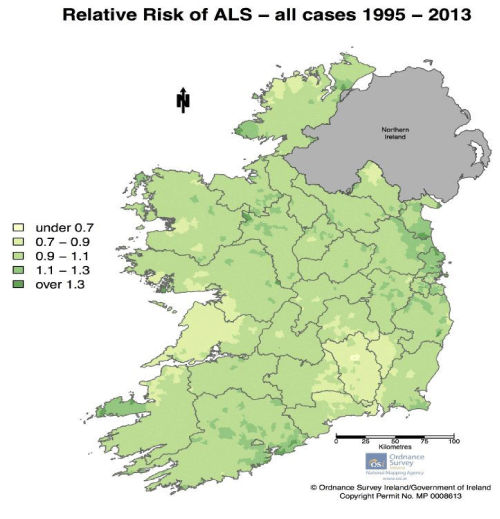
Figure 18: RR ALS ALLA CASES 1995-2013 from reference.
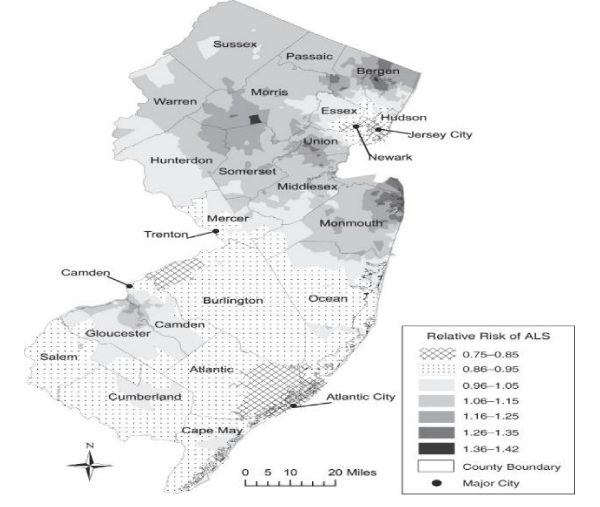
Figure 19: RR of ALS from reference Geographically smoothed relative risk map of amyotrophic lateral sclerosis (ALS) in New Jersey, 2009–2011. The map is based on results from a Bayesian geoadditive model with adjustment for age and sex.
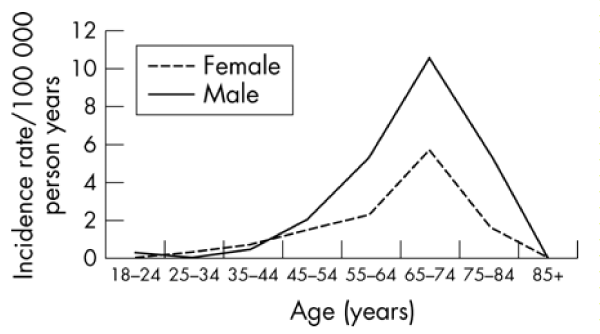
Figure 20: ALS incidence rate.
Figure 21: Incidence of ALS USA.
Figure 22: Prevence – incidence ALS.
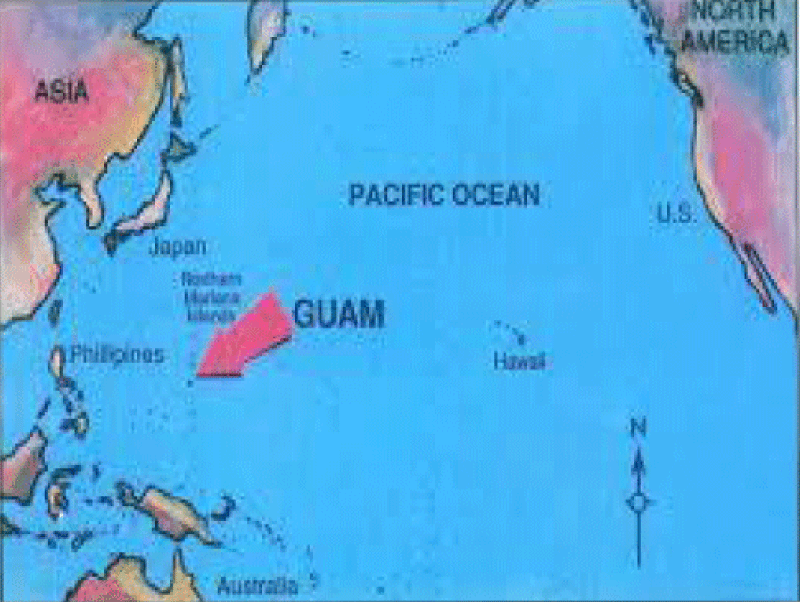
Figure 23: GUAM FOCI OF ALS.
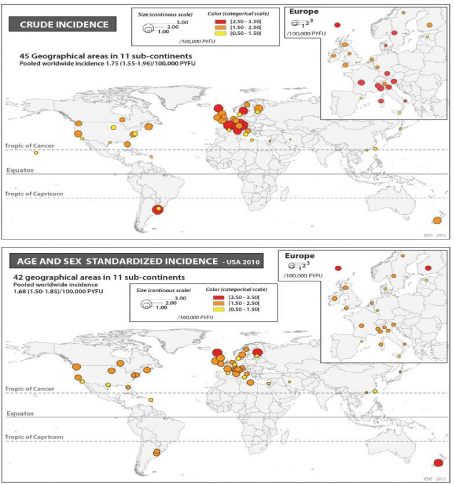
Figure 24: ALS INCIDENCE.
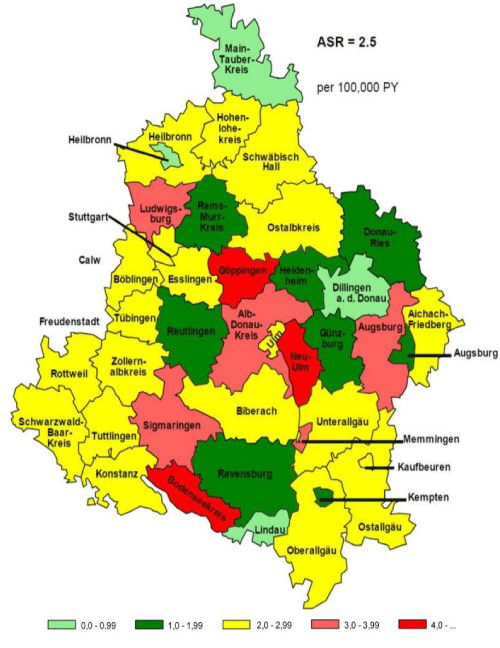
Figure 25: ALS incidence and geographic variation of cases (Form reference).
Figure 26:
Appears to be associated with SES and race” [39].
Experimental project
After this review process is interesting to submit to the researcher an experimental project hypotesys to verify the global conclusion presented:
In order to verify other therapeutic strategies is possible to think to treat neuronal tissue with some Detoxicant or depurative or preventive methods to reduce the global toxic movens produced by the different Plant toxicant substanties.
If the toxic substantia is removed from micro local environment what is the result in progression of some Neuro degenerative disease?
In article Amyotrophic Lateral Sclerosis and Endogenous-Esogenous Toxicological Movens: New Model to Verify Other Pharmacological Strategies. J of Pharmacol& Clin Res. 2018; lusetto et al is reported:
“ALS is a chronic disease and so is related to the progressive damage time related so is possible to think to a in vitro model that compare normal nerve tissue in first fase of disease with other normal nerve tissue not involved in this kind of pathology and observing if local microenvironment contributes in high way to the progression. The same verify this condition in first phases and then advanced phases of disease (progression). In this way is possible to confirm or not soluble factors involved. The same also using the same liquid medium we can observe if present an intrinsic weakness of the nerve tissue affected vs normal tissue. In this experimental project we can show even intrinsic weakness of neurons but also the activity of a soluble factor:
In example 4 IN VITRO experimental sample (animal model)
a) NEURONS SLA AFFECTED + Liquid medium of affected tissue
b) NEURSON SLA AFFECTED + Normal liquid medium
c) NEURONS NOT SLA AFECTED + liquid medium affected tissue
d) NEURONS NOT SLA AFFECTES + normal liquid medium” [69].
The same is interesting verify if using a medical device system is possible to get a more persistence of activity of the measure adopted (pharmacological depurative, detoxicant or with other mechanism that can improve the persistence of action in local spinal cord) [69].
Discussion
Observing table 1 is possible to verify some condition related ASL presentation o(r other neurodegenerative disease):
| Table 1: Related to the reference cited In this work. | |||
| ALS AND RELATED PATHOLOGY | FACTORS INVOLVED | GEOGRAPHIC INVOLVEMENT | |
| AlS GENETIC- SPORADIC | |||
| DIFFUSION | CLUSTER ,GEOGRAFIC PRESENTATION | IN EXAMPLE GUAM FOCI, MUTATION | (21) |
| FIRST DESCRIPTION OF MURO DISEASE (47) | IN YEAR: 1689 | ||
| AVERAGE AGE OF PRESENTATION | 50-65 | ||
| NERUOTOXICITY OF ANNONACEA | PROVED (3) | ||
| ANNONACEA AS CAUSE OF ALS | NOT PROVED | Risk factor | |
| LATIRISM | seed of Lathyrus sativus | ASIA , AFRICA | (22) |
| ELECTRICAL ACTIVITY ELECTROMAGNETIC FIELD |
In mouse : 60 Hz, MF could deteriorate antioxidant defensive system by reactive oxygen species (ROS), other than superoxide radicals (71) | Occupational exposure to ELF-EMF was significantly associated with increased risk of ALS in pooled studies (RR = 1.29, 95%CI = 1.02-1.62), and case-control studies (OR = 1.39, 95%CI = 1.05-1.84) | SOME EVIDENCE Slight but significative association whit high level of exposure See references |
| Electrical injury | Only non progressive damages in more severe shock | Less certain | |
| GENETIC INVOLVEMENT | MUTATIONS IN SOME FORM (59) | SOD1 GEOGRAPHYC DIFFUSION ET OTHER | FAMILIAL FORM Progression of damages |
| Pathogenetic movens | Genetic mutation, oxidative, toxic, inflammatory, cell inclusion? | Uncleared CLEARED ONLY FOR MUTATIONS |
|
| VIRUSES | HERV? | (53) | Transgenic models show motorneuron damages |
| Mitocondrial involvement | Sod1 mutation et other | (59) | |
| OXIDATIVE DAMAGES | |||
| TDP-43 function | TDP-43 functions as a transcriptional repressor and splicing regulator where it is present in ubiquitin-positive, tau and α-synuclein negative inclusions in front temporal lobar degeneration with ubiquitinated inclusions (FTLD-U) and in ALS. , it has been shown to occur in some cases of Lewy body disorders alone or in association with AD (23) | (74) | Progression of damages |
| Astrocite , microglia role | (61) | Neuroinflamation Progression of damages |
|
| NEURONAL intracitoplasmatic INCLUSION | Exposure to annonacin led to phosphorylation and somatodendritic redistribution of tau in a manner similar to MPTP effects (23) | Progression of damage | |
| Well water USE | (52) | CORRELATION WHIT MOTONEURON MORTALITY ? | Nocardia staeroides model BMAA |
| Cyanobacterial β-N-methylamino-l-alanine (BMAA) | INVOLVED? NOT PROVED OR CONTRIBUTORY | Neurotoxin (16) | Controversy relationship whit SOME ALS FORM |
| PESTICIDES | INVOLVED | Showed relationship | |
| SPORTS | FOOTBAL (55), BASEBALL Lou Gehrig disease |
Not OTHER? Basket? | WHY? IRRIGATION PROBLEM? DOPING? SUPPLEMENTARY DIET? DRUGS? |
| Intensive sport physical activity | Not clearly related to the intensity | ||
| Neurotrauma in sports | (73) | Not clearly related | |
| ISCHEMIC ORIGIN | (68) | SOME EVIDENCE | |
| SIZE – DIMENSION OF NEURON | More thickness of sensitive neuron vs motor | ||
| Neurotophic protective factors | VEGF (59) | ||
| HEAVY METALS | Mercury(24) | ||
| Lipophility property of plant toxins involved | (31) | ||
Is possible to easily verify:
A GEOGRAFIC CLUSTER presentation
Environmental factors involved in some cases (toxic plants, BMAA), mercury
A singular genetic diffusion of the pathology (mutations)
Some sports relationship. (Football, baseball)
Electromagnetic field involvement
Intracellular neuronal inclusion (process like other neurodegenerative disease) and other.
Even if a clear relationship between annonacea edibile use and some ASL form is not verified is clear showed the neuro toxicity of this plant.
The geographic limitation of west pacific foci of ALS/PD (GUAM) is another fact to take in consideration.
And the neurotoxic effect seen in some neurological condition like ALS, PD and other is clear. (intracitoplasmatic inclusion).
Annonacea or Lathyrus sativus are proved as neurotroxicological plants. (see ASL and latirism pathology).
This neurotoxic process is a usefull model to deel investigate some neurological pathology. (Brain and spinal cord).
Also rotenone (a natural products of some leguminose plants like Derris elliptica, Derris involuta, Lonchocarpus utilis, Lonchocarpus urucu) is another model of neurotoxicity.
(phytosterols and sterol glucosides in diet and the Lipophylic properties of some plants molecule can accumulate in brain of toxic subtanties).
Is also present and individual variability in toxic responce related the individual genetic profile.(mutation SOD 1 et other) involved in oxidative metabolism.
Other factors that seem to contribute are: Cyanobacterial β-N-methylamino-l-alanine (BMAA) bioproducts that has been suggested as a causative or contributory factor in the development of several neurodegenerative diseases. The cyanobacterial metabolite β-N-methylamino-l-alanine (BMAA), found in many commonly consumed food items, has been implicated in the development of the neurodegenerative diseases ALS /Parkinsonism Dementia Complex (ALS/PDC), (ALS) and Alzheimer’s Disease (AD).
Association was showed by literature with some well water use and related some electromagnetic field exposition.
Neurotoxic signal with apoptosis and other neuronal modification can be seen in many plants derivates models. (latyrism).
Mitochondrial disfunctions, intracellular aggregates and other mechanism contribute in global response.
Related the strange Role of some sports:
-neurotrauma can be etiologic factor or contribute in some subject in ALS presentation?
-Role of dietary supplement, integrators, in sports? (“fruttosio 1,6 bifosfate, surrenal extract cortex, ramified aminoacids, creatin) from artilce: “ Storia del rapporto tra sclerosi laterale amiotrofica e sport: alla ricerca dell’etiopatogenesi sconosciuta Alessandro Cristani [66], Elisa Romagnoli 2006) and about Doping ? And drugs role? Not clearly verified. (MANY TIMES are UNKNOWN DATA the real intake by agonistic players).
Pesticide and herbicide role used in soccer field play a deterniate role? Or water irrigation? (see the particular distribution of cases in central player vs goalkeeper).
According article: Amyotrophic Lateral Sclerosis and Endogenous-Esogenous Toxicological Movens: New Model to Verify Other Pharmacological Strategies. J of Pharmacol “related the body region of onset, mix of upper and lower motoneuron deficits and rate of progression. The endogenous neuro microenvironment in determinate genetic profile is heavily involved with the neuronal damages and strategies that can control or modify it can be useful in preventing progression of some neuro chronic degenerative disease. An exogenous or endogenous toxicology approach (similar to an antidothes approach or depurative strategies) added to the best new pharmaceutical instrument can be a way to be run to protect the motoneuron from a poison like process. The cell death due by apoptosis (free radicals, excitotoxicity, flogosis, immune reactions, toxic exogenous substances and other can be avoided or reduced introducing new depurative strategies (against TOXIC-X Or dangerous micro- or local environment) or other Tanique’s to shift the oxidative damage from the motoneuron to other substances (or other physic procedure, medical devices and other artificial implants to improve global activity). If considered like VECTORS in Physical science the 2 vectors: intrinsic neuronal weakness and the exogenous endogenous toxicologic substances moves in the same direction: a moto neuron damage. Related to this conclusion new pharmacological strategies or high improving in local availability of therapeutic substations can improve clinical outcomes by clinicians. (Better efficiency in pharmacokinetics, BEE pass level, persistence of action, low local toxicity, and other properties, medical devices use, innovative Nano drug delivery systems, alternative way of sub ministration).
A Medicinal chemistry- pharmaceutical and toxicological approach can be the right instrument to be added to the actual therapeutic scenario of this neurodegenerative disease (better pharmacokinetics in BEE pass can be relevant also in other field like oncology- emetology) [69].
Related the caractheristic cluster presentationof some form:
Caller TA et al., “An environmental trigger of sporadic amyotrophic lateral sclerosis (ALS) is supported by geographic disparities in ALS incidence and development of the disease in conjugal couples. This study aims to investigate the incidence of ALS in the Northern New England states of New Hampshire (NH), Vermont (VT), and Maine (ME).
We reviewed medical records and community databases to identify dwelling addresses of 688 patients diagnosed with ALS in 1997-2009 in NH, VT, and ME. We used spatial analysis to identify clusters of census block groups with statistically significant high incidence.
We identified 11 clusters of statistically significant high incidence, each containing 6 or more cases of ALS. These 11 clusters are grouped in 4 distinct regions.
There appear to be areas of significant spatial clustering within Northern New England. Further analysis will be needed to confirm whether there is any correlation between these areas and potential environmental risk factors” [71].
Figure 17: Vulnerability results for each network property and a visualization of critical neurons. (A-C) Sorted vulnerability values (red) for each network.
Conclusion
Some characteristic of ALS disease are very peculiar: environmental factors, mutation geographic diffusion, physical factors, sport involvement, cluster presentation, oxidative methabolism role, viruses, pesticides, well water and cyanobacteria et other.
If geographic implication in some foci is clear (due by some mutation or GUAM FOCI) and in example is clear the association of latirism and seed of Lathyrus sativus, less clear is association of some west pacific form of ALS and annonacea food derivates even if this food behavior is characteristic in some geography zone. (Not in EUROPE, or USA).(Monkeys fed with large doses of the toxic acid from cycads developed neurological impairments).
Is possible to verify the characteristic geographyc diffusion of the Mutation in genes SOD1, gene is TARDBP, which codes for TDP-43, a protein regulating RNA expression and the major component of intracellular inclusions in ALS is another fact to take in consideration.
In many Neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), (ALS), prion diseases are increasingly being realized to have common cellular and molecular mechanisms including protein aggregation and inclusion body formation.
The aggregates usually consist of fibers containing misfolded protein with a beta-sheet conformation, termed amyloid or other products.
Cianobacteria and BMAA, aquatic and terrestrial environments worldwide, well water seems to contribute in this kind of pathology.(In vitro and in vivo experiments also suggest that L-BMAA plays a role in neuropathological processes.) and the biomagnifications we have seen in reported literature can explain the role of concentration of some neuro toxic factors.
Well water usage; bathing habits; food consumption including dietary supplements (to be specified if applicable), and irrigation water tecnique can be factor to be considered? Cluster presentation and sports pattern?
In basket the playground is often syntetic and don t need frequent irrigation. (Goal keeper play at the lateral zone of the field for higher time of median players.
Increased expression of human endogenous retro virus type K (HERV-K): in transgenic models verified mootr neuron damages [53].
Another phenomena is ALS of ischemic origin [68].
Or related to some physical effects:
“Systematic review of the literature on the relationship between ALS and electrical injury indicates that a syndrome of non‐progressive spinal cord damage, often with both LMN and UMN components, is strongly linked to more severe shock, and that the onset of disease is closely related to time from the injury” [37].
But electric field can modify or influence oxidative metabolism?
Why ALS implicated I some sports like football? Neurotrauma, Doping or irrigating problems of the playground, or intensity of phisic movements?
Why are more implied central players vs goal keeper? There is a different exposition OD some toxic agent?
Or are related to a more high energy request due to their role?
Ripetute neuro Trauma seems not play a relevant role in the development of ASL.
Role of Pesticides: in developed countries more used versus not developed countries and this make possible to verify related prevalence of ASL.
But if geographic pattern of diffusion can be a virus like HERV involved or other viruses?
ALS peak age of onset is 55 to 70 years: this is a fact that can be related to an infective process?
Electrical injury? But in 1689 (MURO DIESESE) this factor was not possible.
Exitotoxicity, glutamate, calcium intake, electromagnetic radiations, and mutation, intracellular inclusion are reported in literature.
So result crucial to observe: “motor neurons, as they are post-mitotic cells, are particularly vulnerable to aggregated protein damage since they are unable to reduce it by distributing [aggregates] among daughter cells”.
The GEOGRAPHYC RELATIONSHIP: GUAM foci, IRELAND FRANCE, USA clusters, High prevalence of ALS in Italian cohort of football players seem to show that a diffuse environmental factors can not to be a single cause.
The Characteristic phenomena involving some football player median player more involved then goalkeeper can be related to irrigation or pesticide- herbicide used?
“Heavy physical stress could enhance the production of reactive oxygen species, leading to nucleic acid damage (Julien, 2001), particularly in association with dietary supplements (branched-chain amino acids, BCAAs) and drugs.”
Muro disease described first in a book published in 1689. (Not pesticides related, or electromagnetic force?)
Finally some facts can be observed: Brain os spinal accumulation of some plants toxic substatnties, (lipophilic profileof toxic agent).
Synergic action (cyanobacteria examples) of some fetors, biomagnification of accumulating of toxic substantie.
Mitochondrial effect (sod 1 mutation) and on oxidative metabolism.
Inra- cellular inclusion
Local damages, progression
Astrocites – microglia activation
Apoptosis (final effect)
Genetic profile (influence the metabolic, immunitary of inflammatory response)
But also other physical factor must be taken in consideration in example: “A strong association has been detected between the electric shock or injury and development of ALS (P = 0.003, OR = 6.20, 95% CI = 1.75-21.98)” [27].
Electromagnetic field can affect SOD metabolism?
Is possible to consider not only one single etiological cause but a sort of toxic damage and to consider an UNDER THE CURVE GLOBAL TOXIC MOVENS? (Related to oxidative neuronal metabolism)
And a toxic dose level / response?
Different kind of disease but unic pathologic final effect. When various damages stimulus affect the cell apoptosis is the final effect.
In this work is possible to verify that according literature observed many toxic force can affect motor neuron whit a great reactive gliosis reaction.
But some questions make possible to verify better the problem:
Why this disease involved motor neuron first and not sensitive?
Why neuronal death involve different region of CNS in example ALS, PD, dementia other? (Selectivity of toxicity or different neuronal weakness?)
Why in sports like basket is not present same incidence on ALS then in football? (In basket are used syntetic ground).
In soccer the goalkeeper is on the board of the field, median player in center of the ground for more time.(is irrigation technique related? Or herbicide?)
Biomagnification process of some environmental toxins can influence the process (BMAA), a dosage problem of the neurotoxic subtantia?
Well water use or irrigation can be a model? (nocardia asteroids et other?)
The high lipofilic properties of some plants or bacteria derivates is a clear factor useful in new therapy strategy? And the neurotropism showed by some bacteria in some models can be the same?
What the role of neuron vulnerability? (Interneuron?)
Also the time of exposure is relevant and differ from embrional, neonatal and adult. (Related maturation of CNS?)
SIZE AND METHABOLISM OF MOTOR NEURON VERSUS SENTITIVE PLAY A ROLE?
Can we more efficacy control the gliosis reaction?
For this reason is interesting to submit to researcher to introduce the concept of GLOBAL AREA UNDER THE CURVE related neurotoxic movens related to a specific genetic profile.
If considered this aspect if possible to think in a depurative antidotic strategy in a chronic and progressive disease?
The progression of the disease (ALS) is a direct function of: kind of pathologic damages, time of exposure, level of exposure to toxic/pathologic movens, intrinsic weakness of motor neuron but inverse related to depurative- detoxicant Strategies.
And medical devices (not only drugs) can help to modify the progression acting vs oxidative damages?
A global toxicological approach is the right instrument in this kind of progressive disease.
Clarifications
This works is produced under a pharmaceutical point of view and with no any diagnostic or therapeutic intent, only to produce new research hypotesys.
References
- Escobar-Khondiker M, Höllerhage M, Muriel MP, Champy P, Bach A, et al. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J Neurosci. 2007; 27: 7827-7837. Ref.: https://goo.gl/3cFDm8
- N Bonneau, J Le Ven, I Schmitz-Afonso, V Guérineau, I Bajin ba Ndob, et al. Annonaceous acetogenins as environmental neurotoxins: Human exposure from edible Annona fruits. Planta Med. 2012; 78 - PH25. Ref.: https://goo.gl/fdZdir
- Lannuzel A, Michel PP, Caparros-Lefebvre D, Abaul J, Hocquemiller R, et al. Toxicity of Annonaceae for dopaminergic neurons: Potential role in atypical parkinsonism in Guadeloupe. Mov Disord. 2002;17: 84-90. Ref.: https://goo.gl/NhQaGs
- Höllerhage M, Rösler TW, Berjas M, Luo R, Tran K, et al. Neurotoxicity of Dietary Supplements from Annonaceae Species. Int J Toxicol. 2015; 34: 543-550. Ref.: https://goo.gl/3Dn27B
- Zafra-Polo MC, González MC, Estornell E, Sahpaz S, Cortes D. Acetogenins from Annonaceae, inhibitors of mitochondrial complex I. Phytochemistry. 1996; 42: 253-271. Ref.: https://goo.gl/Sdmqi2
- P. N'Gouemo, B. Koudogbo, H. Pambou Tchivounda, C. Akono‐Nguema, Minko M. Etoua
- Effects of ethanol extract of Annona muricata on pentylenetetrazol‐induced convulsive seizures in mice. Short Communication. 1998. Ref.: https://goo.gl/Dgr8fR
- Moghadamtousi SZ, Rouhollahi E, Hajrezaie M, Karimian H, Abdulla MA, et al. Annona muricata leaves accelerate wound healing in rats via involvement of Hsp70 and antioxidant defence. Int J Surg. 2015; 18: 110-117. Ref.: https://goo.gl/S8fkmi
- Chen Y, Chen JW, Zhai JH, Wang Y, Wang SL, et al. Antitumor activity and toxicity relationship of annonaceous acetogenins. Food Chem Toxicol. 2013; 58: 394-400. Ref.: https://goo.gl/7jsbTN
- Ribeiro Ldo P, Zanardi OZ, Vendramim JD, Yamamoto PT. Comparative toxicity of an acetogenin-based extract and commercial pesticides against citrus red mite. Exp Appl Acarol. 2014; 64: 87-98. Ref.: https://goo.gl/vnpnc7
- Levine RA, Richards KM, Tran K, Luo R, Thomas AL, et al. Determination of Neurotoxic Acetogenins in Pawpaw (Asimina triloba) Fruit by LC-HRMS. J Agric Food Chem. 2015; 63: 1053-1056. Ref.: https://goo.gl/WBmX6q
- Jiang Z, Wang W, Perry G, Zhu X, Wang X. Mitochondrial dynamic abnormalities in amyotrophic lateral sclerosis. Transl Neurodegener. 2015; 4: 14. Ref.: https://goo.gl/vxJowD
- Ravindranath V. Neurolathyrism: mitochondrial dysfunction in excitotoxicity mediated by L-beta-oxalyl aminoalanine. Neurochem Int. 2002; 40: 505-509. Ref.: https://goo.gl/AueXgQ
- Potts LF, Luzzio FA, Smith SC, Hetman M, Champy P, et al. Annonacin in Asimina triloba fruit: Implication for neurotoxicity. Neurotoxicology. 2012; 33: 53-58. Ref.: https://goo.gl/qi3SpF
- Panov A, Dikalov S, Shalbuyeva N, Taylor G, Sherer T, et al. Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J Biol Chem. 2005; 280: 42026-42035. Ref.: https://goo.gl/yYvYvH
- Shen WB, McDowell KA, Siebert AA, Clark SM, Dugger NV, et al. Environmental neurotoxin-induced progressive model of parkinsonism in rats. Ann Neurol. 2010; 68: 70-80. Ref.: https://goo.gl/3M1CtV
- Scott LL, Downing TG. B-N-Methylamino-l-alanine (BMAA) Toxicity Is Gender and Exposure-Age Dependent in Rats. Toxins (Basel). 2018; 10: 16. Ref.: https://goo.gl/69V7GM
- Uversky VN. Neurotoxicant-induced animal models of Parkinson's disease: understanding the role of rotenone, maneb and paraquat in neurodegeneration. Cell Tissue Res. 2004; 318: 225-241. Ref.: https://goo.gl/3Erqvj
- de Pedro N, Cautain B, Melguizo A, Vicente F, Genilloud O, Peláez F, et al. Mitochondrial complex I inhibitors, acetogenins, induce HepG2 cell death through the induction of the complete apoptotic mitochondrial pathway. J Bioenerg Biomembr. 2013; 45: 153-164. Ref.: https://goo.gl/YqcVof
- Degli Esposti M, Ghelli A, Ratta M, Cortes D, Estornell E. Natural substances (acetogenins) from the family Annonaceae are powerful inhibitors of mitochondrial NADH dehydrogenase (Complex I). Biochem J. 1994; 301: 161-167. Ref.: https://goo.gl/gqXFpE
- Segura Aguilar J, Kostrzewa RM. Neurotoxins and neurotoxic species implicated in neurodegeneration. Neurotox Res. 2004; 6: 615-630. Ref.: https://goo.gl/h1K7Sc
- Caparros-Lefebvre D, Steele J, Kotake Y, Ohta S. Geographic isolates of atypical Parkinsonism and tauopathy in the tropics: possible synergy of neurotoxins. Mov Disord. 2006; 21: 1769-1771. Ref.: https://goo.gl/twX6pZ
- Khandare AL, Kumar RH, Meshram II, Arlappa N, Laxmaiah A, et al. Current scenario of consumption of Lathyrus sativus and lathyrism in three districts of Chhattisgarh State, India. Toxicon. 2018; 150: 228-234. Ref.: https://goo.gl/Kf6kGC
- Salama M, Arias-Carrión O. Natural toxins implicated in the development of Parkinson’s disease. Ther Adv Neurol Disord. 2011; 4: 361-373. Ref.: https://goo.gl/fo1KA7
- Bozzoni V, Pansarasa O, Diamanti L, Nosari G, Cereda C. Amyotrophic lateral sclerosis and environmental factors. Funct Neurol. 2016; 31: 7-19. Ref.: https://goo.gl/UpPN7K
- Pan-Montojo F, Reichmann H. Considerations on the role of environmental toxins in idiopathic Parkinson’s disease pathophysiology. Transl Neurodegener. 2014; 3: 10. Ref.: https://goo.gl/EJ9URY
- Rafael H, David JO, Vilca AS. Etiology and treatment of amyotrophic lateral sclerosis. Am J Neurodegener Dis. 2017; 6: 1-8. Ref.: https://goo.gl/rtZ4Ra
- Das K, Nag C, Ghosh M. Familial, Environmental, and Occupational Risk Factors in Development of Amyotrophic Lateral Sclerosis. N Am J Med Sci. 2012; 4: 350-355. Ref.: https://goo.gl/s1hzKe
- Malek AM, Barchowsky A, Bowser R, Youk A, Talbott EO. Pesticide exposure as a risk factor for amyotrophic lateral sclerosis: a meta-analysis of epidemiological studies: pesticide exposure as a risk factor for ALS. Environ Res. 2012; 117: 112-119. Ref.: https://goo.gl/WU5nH4
- Zhou H, Chen G, Chen C, Yu Y, Xu Z. Association between extremely low-frequency electromagnetic fields occupations and amyotrophic lateral sclerosis: a meta-analysis. PLoS One. 2012; 7: e48354. Ref.: https://goo.gl/HfDCt4
- Banack SA, Cox PA. Biomagnification of cycad neurotoxins in flying foxes: implications for ALS-PDC in Guam. Neurology. 2003; 61: 387-389. Ref.: https://goo.gl/Y6kXf2
- Cox PA, Banack SA, Murch SJ. Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam. Proc Natl Acad Sci U S A. 2003; 100: 13380-13383. Ref.: https://goo.gl/Xm4YVK
- Ingre C, Roos PM, Piehl F, Kamel F, Fang F. Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol. 2015; 7: 181-193. Ref.: https://goo.gl/Lgk4yL
- Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol. 2010; 67: 739-748. Ref.: https://goo.gl/v2VJqB
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004; 10: S10-S17. Ref.: https://goo.gl/JoNjK7
- Huss A, Spoerri A, Egger M, Kromhout H, Vermeulen R, et al. Occupational exposure to magnetic fields and electric shocks and risk of ALS: the Swiss National Cohort. Amyotroph Lateral Scler Frontotemporal Degener. 2015; 16: 80-85. Ref.: https://goo.gl/W5AeYm
- Martin S, Al Khleifat A, Al-Chalabi A. What causes amyotrophic lateral sclerosis? F1000Res. 2017; 6: 371. Ref.: https://goo.gl/KJTgiH
- Abhinav K, Al-Chalabi A, Hortobagyi T, Leigh PN. Electrical injury and amyotrophic lateral sclerosis: a systematic review of the literature. J Neurol Neurosurg Psychiatry. 2007; 78: 450-453. Ref.: https://goo.gl/8TSPgf
- Delzor A, Couratier P, Boumédiène F, Nicol M, Druet-Cabanac M, et al. Searching for a link between the L-BMAA neurotoxin and amyotrophic lateral sclerosis: a study protocol of the French BMAALS programme. BMJ Open. 2014; 4: e005528. Ref.: https://goo.gl/mGcWPh
- Henry KA, Fagliano J, Jordan HM, Rechtman L, Kaye WE. Geographic Variation of Amyotrophic Lateral Sclerosis Incidence in New Jersey, 2009–2011. Am J Epidemiol. 2015; 182: 512-519. Ref.: https://goo.gl/mD75DD
- Rooney J, Heverin M, Vajda A, Crampsie A, Tobin K, et al. An Exploratory Spatial Analysis of ALS Incidence in Ireland over 17.5 Years (1995 – July 2013). PLoS One. 2014; 9: e96556. Ref.: https://goo.gl/jCeEyt
- Logroscino G, Piccininni M. Amyotrophic Lateral Sclerosis Descriptive Epidemiology: The Origin of Geographic Difference. Neuroepidemiology. 2019; 52: 93-103. Ref.: https://goo.gl/2hDwXo
- Scott KM, Abhinav K, Stanton BR, Johnston C, Turner MR, et al. Geographical clustering of amyotrophic lateral sclerosis in South-East England: a population study. Neuroepidemiology. 2009; 32: 81-88. Ref.: https://goo.gl/ZrVzPN
- Uenal H, Rosenbohm A, Kufeldt J, Weydt P, Goder K, et al. Incidence and Geographical Variation of Amyotrophic Lateral Sclerosis (ALS) in Southern Germany – Completeness of the ALS Registry Swabia. PLoS One. 2014; 9: e93932. Ref.: https://goo.gl/5hjGV4
- Kokubo Y, Kuzuhara S, Narita Y. Geographical distribution of amyotrophic lateral sclerosis with neurofibrillary tangles in the Kii Peninsula of Japan. J Neurol. 2000; 247: 850-852. Ref.: https://goo.gl/oqGkV8
- Virginia Bozzoni, Orietta Pansarasa, Luca Diamanti, Guido Nosari, Cristina Cereda, et al. Amyotrophic lateral sclerosis and environmental factors. Funct Neurol. 2016; 31: 7-19. Ref.: https://goo.gl/WpMiMG
- Yu Y, Su FC, Callaghan BC, Goutman SA, Batterman SA, et al. Environmental Risk Factors and Amyotrophic Lateral Sclerosis (ALS): A Case-Control Study of ALS in Michigan. PLoS One. 2014; 9: e101186. Ref.: https://goo.gl/USCX5B
- Kuzuhara S. [ALS-parkinsonism-dementia complex of the Kii peninsula of Japan (Muro disease). Historical review, epidemiology and concept]. Rinsho Shinkeigaku. 2007; 47: 962-965. Ref.: https://goo.gl/dGrJ61
- Zarei S, Carr K, Reiley L, Diaz K, Guerra O, et al. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. 2015; 6: 171. Ref.: https://goo.gl/WAWsUw
- Ludolph AC, Hugon J, Dwivedi MP, Schaumburg HH, Spencer PS. Studies on the aetiology and pathogenesis of motor neuron diseases. 1. Lathyrism: clinical findings in established cases. Brain. 1987; 110: 149-465. Ref.: https://goo.gl/Z7Q7KU
- Khandare AL, Babu JJ, Ankulu M, Aparna N, Shirfule A, et al. Grass pea consumption & present scenario of neurolathyrism in Maharashtra State of India. Indian J Med Res. 2014; 140: 96-101. Ref.: https://goo.gl/bW4wgx
- Uccelli R, Binazzi A, Altavista P, Belli S, Comba P, et al. Geographic distribution of amyotrophic lateral sclerosis through motor neuron disease mortality data. Eur J Epidemiol. 2007; 22: 781-790. Ref.: https://goo.gl/548yma
- Schwartz GG, Rundquist BC, Simon IJ, Swartz SE. Geographic distributions of motor neuron disease mortality and well water use in U.S. counties. Amyotroph Lateral Scler Frontotemporal Degener. 2017; 18: 279-283. Ref.: https://goo.gl/uDefbU
- Li W, Lee MH, Henderson L, Tyagi R, Bachani M, et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med. 2015; 7: 307ra153. Ref.: https://goo.gl/FbY7so
- Chio A, Calvo A, Dossena M, Ghiglione P, Mutani R, et al. ALS in Italian professional soccer players: the risk is still present and could be soccer-specific. Amyotroph Lateral Scler. 2009; 10: 205-209. Ref.: https://goo.gl/BdqgVD
- Chiò A, Benzi G, Dossena M, Mutani R, Mora G. Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain. 2005; 128: 472-476. Ref.: https://goo.gl/yz7Ubm
- Belli S, Vanacore N. Proportionate mortality of Italian soccer players: is amyotrophic lateral sclerosis an occupational disease? Eur J Epidemiol. 2005; 20: 237-242. Ref.: https://goo.gl/r2Zfg3
- Delzor A, Couratier P, Boumédiène F, Nicol M, Druet-Cabanac M, et al. Searching for a link between the L-BMAA neurotoxin and amyotrophic lateral sclerosis: a study protocol of the French BMAALS programme. BMJ Open. 2014; 4: e005528. Ref.: https://goo.gl/RKPkSP
- Manzano GM, Giuliano LM, Nóbrega JA. A brief historical note on the classification of nerve fibers Arq Neuropsiquiatr. 2008; 66: 117-119. Ref.: https://goo.gl/8ZcD7C
- Tapia R. Cellular and molecular mechanisms of motor neuron death in amyotrophic lateral sclerosis: a perspective. Front Cell Neurosci. 2014; 8: 241. Ref.: https://goo.gl/bLTZqR
- Bélanger M, Allaman I, Magistretti PJ. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011; 14: 724-738. Ref.: https://goo.gl/6WLzWp
- Falkowska A, Gutowska I, Goschorska M, Nowacki P, Chlubek D, et al. Energy Metabolism of the Brain, Including the Cooperation between Astrocytes and Neurons, Especially in the Context of Glycogen Metabolism. Int J Mol Sci. 2015; 16: 25959-25981. Ref.: https://goo.gl/smjsmd
- Bertamini M, Marzani B, Guarneri R, Guarneri P, Bigini P, et al. Mitochondrial oxidative metabolism in motor neuron degeneration (mnd) mouse central nervous system. Eur J Neurosci. 2002; 16: 2291-2296. Ref.: https://goo.gl/KVDq6F
- Tefera TW, Borges K. Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments. Front Neurosci. 2016; 10: 611. Ref.: https://goo.gl/EhcDe5
- Belli S, Vanacore N. Proportionate mortality of Italian soccer players: is amyotrophic lateral sclerosis an occupational disease? Eur J Epidemiol. 2005; 20: 237-242. Ref.: https://goo.gl/kMmb95
- Malek AM, Barchowsky A, Bowser R, Youk A, Talbott EO. Pesticide exposure as a risk factor for amyotrophic lateral sclerosis: a meta-analysis of epidemiological studies: pesticide exposure as a risk factor for ALS. Environ Res. 2012; 117: 112-119. Ref.: https://goo.gl/bA3Xie
- Alessandro Cristani, Elisa Romagnoli. Storia del rapporto tra sclerosi laterale amiotrofica e sport: alla ricerca dell’etiopatogenesi sconosciuta. Recenti Progressi Medicina. 2006. Ref.: https://goo.gl/33y53X
- Feddermann-Demont N, Junge A, Weber KP, Weller M, Dvořák J, et al. Prevalence of potential sports‐associated risk factors in Swiss amyotrophic lateral sclerosis patients. Brain Behav. 2017; 7: e00630. Ref.: https://goo.gl/ikToDA
- Vargas MI, Gariani J, Sztajzel R, Barnaure-Nachbar I, Delattre BM, et al. Spinal Cord Ischemia: Practical Imaging Tips, Pearls, and Pitfalls. AJNR Am J Neuroradiol. 2015; 36: 825-830. Ref.: https://goo.gl/n8L7zd
- Mauro Luisetto , Behzad N-A, Nilesh MM, Ghulam RM, Ram KS, et al. Amyotrophic Lateral Sclerosis and Endogenous-Esogenous Toxicological Movens: New Model to Verify Other Pharmacological Strategies. Arch Pathol Clin Res. 2018; 2: 28-48. Ref.: https://goo.gl/QpkZd6
- Kim S, Kim H, Kralik JD, Jeong J. Vulnerability-Based Critical Neurons, Synapses, and Pathways in the Caenorhabditis elegans Connectome. PLoS Comput Biol. 2016; 12: e1005084. Ref.: https://goo.gl/HedD1H
- Caller TA, Chipman JW, Field NC, Stommel EW. Spatial analysis of amyotrophic lateral sclerosis in Northern New England, USA, 1997-2009. Muscle Nerve. 2013; 48: 235-241. Ref.: https://goo.gl/cSWyYs
- Lee BC, Johng HM, Lim JK, Jeong JH, Baik KY, et al. Effects of extremely low frequency magnetic field on the antioxidant defense system in mouse brain: a chemiluminescence study. J Photochem Photobiol B. 2004; 73: 43-48. Ref.: https://goo.gl/bsRTqj
- Piazza O, Sirén AL, Ehrenreich H. Soccer, neurotrauma and amyotrophic lateral sclerosis: is there a connection? Curr Med Res Opin. 2004; 20: 505-508. Ref.: https://goo.gl/X5g9Zd
- Scotter EL, Chen HJ, Shaw CE. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics. 2015; 12: 352-363. Ref.: https://goo.gl/NkJD6Q
- Sweeney P, Park H, Baumann M, Dunlop J, Frydman J, et al. Protein misfolding in neurodegenerative diseases: implications and strategie. Transl Neurodegener. 2017; 6: 6. Ref.: https://goo.gl/4RdYnD